








Pediatric Adrenoleukodystrophy (ALD).pptx
- 1. Pediatric Adrenoleukodystro phy (ALD) Understanding, diagnosing, and treating genetic disorder affecting children. by Dr. Omondi Obudho Paediatrics Resident II Maseno University
- 2. Introduction to ALD 1 Genetic Disorder X-linked disorder associated with the accumulation of saturated VLCFAs and a progressive dysfunction of the adrenal cortex and nervous system. It is the most common peroxisomal disorder. 2 Peroxisomal Disorder It is a peroxisomal disorder characterized by the abnormal metabolism of very long-chain fatty acids (VLCFAs) due to mutations in the ABCD1 gene 3 Peroxisomes The peroxisome is a subcellular organelle containing enzymes involved in production and decomposition of hydrogen peroxide and others in lipid and amino acid metabolism, surrounded by a single membrane
- 3. Etiology of ALD 1 Genetic Basis X-linked recessive inheritance pattern. 2 ABCD1 Gene The defective gene (ABCD1) codes for a peroxisomal membrane protein (ALDP, the ALD protein). 3 Mechanism The mechanism appears to be a disruption of transport of saturated fatty acids into the peroxisome, with resultant continued elongation of progressively longer fatty acids.
- 4. Epidemilogy of ALD 1 Incidence Incidence in males is 1 in 21,000, and the combined incidence In males and heterozygous females in the general population is 1 in 17,000. 2 Race All races are affected but the disease incidence is higher in patients of Latino or African descent 3 Age of Onset Presents between ages 4 and 10, with rapid progression. Milder form that appears during adolescence or adulthood, with slower progression.
- 5. Published studies in Africa 1 Management of X-linked ALD in Morocco This study outlines the first program in Morocco for diagnosing, treating, and monitoring ALD. Over five years, researchers identified eight families with 16 patients, detailing clinical presentations such as primary adrenal insufficiency and behavioral changes. (F. Z. Madani Benjelloun et al, 2017) 2 Clinical Profile and Diagnostic Challenges in Sudanese Children Conducted over 14 years, this research describes the clinical presentation, potential causes, and diagnostic challenges of primary adrenal insufficiency in Sudanese children. Among the findings, seven cases of ALD were identified, highlighting the difficulties in diagnosis and management within resource-limited settings. (S.A. Musa et al, 2021)
- 6. Pathophysiology VLCFA Accumulation Due to defective ALDP, VLCFAs cannot be properly transported into peroxisomes, leading to their accumulation in the body. Elevated VLCFA levels are toxic to cells, particularly affecting myelin, the protective sheath around nerve fibers in the CNS Demyelination Accumulation of VLCFAs leads to progressive demyelination, this disruption severely impairs the conduction of nerve impulses, resulting in neurological symptoms. Inflammatory response The breakdown of myelin triggers an inflammatory response in the brain, which exacerbates the damage. The immune system's attack on the myelin leads to further neurological decline. Genetic mutation Mutations are in the ABCD1 gene, which encodes ALDP, which is involved in transporting VLCFAs into peroxisomes for degradation. Adrenal insufficiency: VLCFAs also accumulate in the adrenal cortex, impairing its ability to produce adrenal hormones such as cortisol and aldosterone. This results in adrenal insufficiency, leading to symptoms such as fatigue, weight loss, and skin changes.
- 7. Types of ALD Type Onset Key Features Cerebral ALD Childhood Rapid neurological decline Adrenomyeloneuropathy Adulthood Slower progression, spinal cord involvement Addison's-only Variable Adrenal insufficiency without neurological symptoms
- 8. Pediatric ALD Symptoms Childhood Cerebral Form Ages 4 & 8. Mainly is developmental regression, characterized by progressive sensory and severe neurological deficits, leading to significant disability, coma, and eventually death. Adolescent ALD Symptoms between ages 10 and 21, slower progress. Approximately 10% of patients present acutely with status epilepticus, adrenal crisis, acute encephalopathy, or coma Addison-only Of male patients with Addison disease, 25% may have the biochemical defectof ALD. Development is usually normal in the 1st 3-4 yr of life.
- 9. Pediatric ALD Symptoms Adrenomyeloneuropathy First manifests in late adolescence as a progressive paraparesis caused by long tract degeneration in the spinal cord. About half the affected men also have involvement of the cerebral white matter Neonatal adrenoleukodystrophy Seizures, hypotonia, and hearing dysfunction vision loss, cataracts, and optic nerve dysplasia Jaundice and hepatomegaly Failure to thrive and facial dysmorphism (such as hypertelorism and flat midface)
- 10. Diagnosing ALD Blood Tests High VLCFA levels indicate ALD presence. plasma, RBCs, or cultured skin fibroblasts. MRI Scans Reveal characteristic white matter changes. Genetic Testing Confirms ABCD1 gene mutation. Newborn Screening Early identification enables prompt intervention. Impaired Adrenal Function More than 85% of patients with the childhood form of ALD have elevated levels of ACTH in plasma and a subnormal rise of cortisol levels in plasma after IV injection of 250 ╬╝g of ACTH (Cortrosyn).
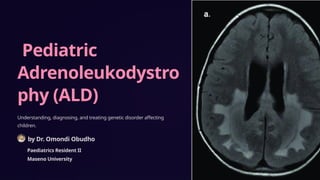
- 11. Neuroimaging in ALD MRI Scans Patients with childhood cerebral or adolescent ALD have characteristic white matter lesions on MRI. In 80% of patients the lesions are symmetric and involve the splenium of the corpus callosum and periventricular white matter in the posterior parietal and occipital lobes. Many will show a garland of contrast enhancement adjacent and anterior to the posterior hypodense lesions
- 12. Treatment Options HSCT Hematopoietic stem cell transplantation halts disease progression. Gene Therapy Emerging treatment correcting faulty ABCD1 gene. Supportive Care Physical, occupational, speech therapies maintain function. Dietary Management Low VLCFA diet may slow disease progression. LorenzoŌĆÖs Oil Therapy LorenzoŌĆÖs oil (4 : 1 mixture of glyceryl trioleate and glyceryl trierucate) combined with a dietary regimen has been under investigation to prevent the development of various aspects of ALD.
- 13. Emerging Therapies 1 Gene Therapy Trials Lentiviral vector therapy shows promise. 2 CRISPR Research Gene editing potential for ABCD1 correction. 3 Drug Development New compounds targeting VLCFA metabolism.
- 14. Future Prospects Advanced Gene Therapy Personalized treatments based on genetic profile. AI-Assisted Diagnosis Early detection through advanced imaging and data analysis. Improved Support Comprehensive care networks for patients and families.