
















01.4.pssm theory
- 1. Jacques.van.Helden@ulb.ac.be Universit¨¦ Libre de Bruxelles, Belgique Laboratoire de Bioinformatique des G¨¦nomes et des R¨¦seaux (BiGRe) http://www.bigre.ulb.ac.be/ 1 Position-specific scoring matrices (PSSM) Regulatory sequence analysis
- 2. 2 . Binding sites for the yeast Pho4p transcription factor (Source : Oshima et al. Gene 179, 1996; 171-177) Alignment of transcription factor binding sites Gene Site Name Sequence Affinity PHO5 UASp2 ---aCtCaCACACGTGGGACTAGC- high PHO84 Site D ---TTTCCAGCACGTGGGGCGGA-- high PHO81 UAS ----TTATGGCACGTGCGAATAA-- high PHO8 Proximal GTGATCGCTGCACGTGGCCCGA--- high group 1 consensus ---------gCACGTGgg------- high PHO5 UASp1 --TAAATTAGCACGTTTTCGC---- medium PHO84 Site E ----AATACGCACGTTTTTAATCTA medium group 2 consensus --------cgCACGTTtt------- medium Degenerate consensus ---------GCACGTKKk------- high-med Non-binding sites PHO5 UASp3 --TAATTTGGCATGTGCGATCTC-- No binding PHO84 Site C -----ACGTCCACGTGGAACTAT-- No binding PHO84 Site A -----TTTATCACGTGACACTTTTT No binding PHO84 Site B -----TTACGCACGTTGGTGCTG-- No binding PHO8 Distal ---TTACCCGCACGCTTAATAT--- No binding IUPAC ambiguous nucleotide code A A Adenine C C Cy tosine G G Guanine T T Thy mine R A or G puRine Y C or T pYrimidine W A or T Weak hy drogen bonding S G or C Strong hy drogen bonding M A or C aMino group at common position K G or T Keto group at common position H A, C or T not G B G, C or T not A V G, A, C not T D G, A or T not C N G, A, C or T aNy
- 3. Jacques.van.Helden@ulb.ac.be Universit¨¦ Libre de Bruxelles, Belgique Laboratoire de Bioinformatique des G¨¦nomes et des R¨¦seaux (BiGRe) http://www.bigre.ulb.ac.be/ 3 From alignments to weights Regulatory sequence analysis
- 4. 4 Sequence logo Tom SchneiderˇŻs sequence logo (generated with Web Logo http://weblogo.berkeley.edu/logo.cgi) Count matrix (TRANSFAC matrix F$PHO4_01) Residueposition 1 2 3 4 5 6 7 8 9 10 11 12 A 1 3 2 0 8 0 0 0 0 0 1 2 C 2 2 3 8 0 8 0 0 0 2 0 2 G 1 2 3 0 0 0 8 0 5 4 5 2 T 4 1 0 0 0 0 0 8 3 2 2 2 Sum 8 8 8 8 8 8 8 8 8 8 8 8
- 5. 5 Frequency matrix Pos 1 2 3 4 5 6 7 8 9 10 11 12 A 0.13 0.38 0.25 0.00 1.00 0.00 0.00 0.00 0.00 0.00 0.13 0.25 C 0.25 0.25 0.38 1.00 0.00 1.00 0.00 0.00 0.00 0.25 0.00 0.25 G 0.13 0.25 0.38 0.00 0.00 0.00 1.00 0.00 0.63 0.50 0.63 0.25 T 0.50 0.13 0.00 0.00 0.00 0.00 0.00 1.00 0.38 0.25 0.25 0.25 Sum 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 A alphabet size (=4) ni,j, occurrences of residue i at position j pi prior residue probability for residue i fi,j relative frequency of residue i at position j Reference: Hertz (1999). Bioinformatics 15:563-577. ! fi, j = ni, j ni, j i=1 A "
- 6. 6 Corrected frequency matrix P r Pos 1 2 3 4 5 6 7 8 9 10 11 12 A 0.15 0.37 0.26 0.04 0.93 0.04 0.04 0.04 0.04 0.04 0.15 0.26 C 0.24 0.24 0.35 0.91 0.02 0.91 0.02 0.02 0.02 0.24 0.02 0.24 G 0.13 0.24 0.35 0.02 0.02 0.02 0.91 0.02 0.58 0.46 0.58 0.24 T 0.48 0.15 0.04 0.04 0.04 0.04 0.04 0.93 0.37 0.26 0.26 0.26 Sum 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 A alphabet size (=4) ni,j, occurrences of residue i at position j pi prior residue probability for residue i fi,j relative frequency of residue i at position j k pseudo weight (arbitrary, 1 in this case) f'i,j corrected frequency of residue i at position j Reference: Hertz (1999). Bioinformatics 15:563-577. ! fi, j ' = ni, j + k /A ni, j i=1 A " + k ! fi, j ' = ni, j + pik ni, j i=1 A " + k 1st option: identically distributed pseudo-weight 2nd option: pseudo-weight distributed according to residue priors
- 7. 7 Weight matrix (Bernoulli model) Prior Pos 1 2 3 4 5 6 7 8 9 10 11 12 0.325 A -0.79 0.13 -0.23 -2.20 1.05 -2.20 -2.20 -2.20 -2.20 -2.20 -0.79 -0.23 0.175 C 0.32 0.32 0.70 1.65 -2.20 1.65 -2.20 -2.20 -2.20 0.32 -2.20 0.32 0.175 G -0.29 0.32 0.70 -2.20 -2.20 -2.20 1.65 -2.20 1.19 0.97 1.19 0.32 0.325 T 0.39 -0.79 -2.20 -2.20 -2.20 -2.20 -2.20 1.05 0.13 -0.23 -0.23 -0.23 1.000 Sum -0.37 -0.02 -1.02 -4.94 -5.55 -4.94 -4.94 -5.55 -3.08 -1.13 -2.03 0.19 A alphabet size (=4) ni,j, occurrences of residue i at position j pi prior residue probability for residue i fi,j relative frequency of residue i at position j k pseudo weight (arbitrary, 1 in this case) f'i,j corrected frequency of residue i at position j Wi,j weight of residue i at position j ! Wi, j = ln fi, j ' pi " # $ % & ' ! fi, j ' = ni, j + pik nr, j r=1 A " + k Reference: Hertz (1999). Bioinformatics 15:563-577. The use of a weight matrix relies on Bernoulli assumption If we assume, for the background model, an independent succession of nucleotides (Bernoulli model), the weight WS of a sequence segment S is simply the sum of weights of the nucleotides at successive positions of the matrix (Wi,j). In this case, it is convenient to convert the PSSM into a weight matrix, which can then be used to assign a score to each position of a given sequence.
- 8. 8 Properties of the weight function ! Wi, j = ln fi, j ' pi " # $ % & ' ! fi, j ' = ni, j + pik ni, j i=1 A " + k fi, j ' i=1 A " =1 ? The weight is ? positive when fˇŻi,j > pi (favourable positions for the binding of the transcription factor) ? negative when fˇŻi,j < pi (unfavourable positions)
- 9. Jacques.van.Helden@ulb.ac.be Universit¨¦ Libre de Bruxelles, Belgique Laboratoire de Bioinformatique des G¨¦nomes et des R¨¦seaux (BiGRe) http://www.bigre.ulb.ac.be/ 9 Information content Regulatory sequence analysis
- 10. 10 Shannon uncertainty ? Shannon uncertainty ? Hs(j): uncertainty of a column of a PSSM ? Hg: uncertainty of the background (e.g. a genome) ? Special cases of uncertainty (for a 4 letter alphabet) ? min(H)=0 ? No uncertainty at all: the nucleotide is completely specified (e.g. p={1,0,0,0}) ? H=1 ? Uncertainty between two letters (e.g. p={0.5,0,0,0.5}) ? max(H) = 2 (Complete uncertainty) ? One bit of information is required to specify the choice between each alternative (e.g. p={0.25,0.25,0.25,0.25}). ? Two bits are required to specify a letter in a 4- letter alphabet. ? Rseq ? Schneider (1986) defines an information content based on ShannonˇŻs uncertainty. ? R* seq ? For skewed genomes (i.e. unequal residue probabilities), Schneider recommends an alternative formula for the information content . This is the formula that is nowadays used. Adapted from Schneider (1986) ! Hs j( )= " fi, j log2( fi, j ) i=1 A # Hg = " pi log2(pi) i=1 A # Rseq j( ) = Hg " Hs j( ) Rseq = Rseq j( ) j=1 w # Rseq * j( ) = fi, j log2 fi, j pi $ % & ' ( ) i=1 A # Rseq * = Rseq * j( ) j=1 w #
- 11. 11 Schneider logos ? Schneider (1990) proposes a graphical representation based on his previous entropy (H) for representing the importance of each residue at each position of an alignment. He provides a new formula for Rseq ? Hs(j) uncertainty of column j ? Rseq(j) ˇ°information contentˇ± of column j (beware, this de?nition differs from HertzˇŻ information content) ? e(n) correction for small samples (pseudo-weight) ? Remarks ? This information content does not include any correction for the prior residue probabilities (pi) ? This information content is expressed in bits. ? Boundaries ? min(Rseq)=0 equiprobable residues ? max(Rseq)=2 perfect conservation of 1 residue with a pseudo-weight of 0, ? Sequence logos can be generated from aligned sequences on the Weblogo server ? http://weblogo.berkeley.edu/ ! Hs j( )= " fij log2( fij ) i=1 A # Rseq j( ) = 2 " Hs j( )+ e n( ) hij = fijRseq ( j) Pho4p binding motif
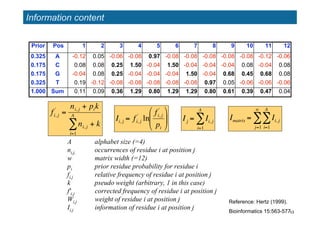
- 13. 13 Information content Prior Pos 1 2 3 4 5 6 7 8 9 10 11 12 0.325 A -0.12 0.05 -0.06 -0.08 0.97 -0.08 -0.08 -0.08 -0.08 -0.08 -0.12 -0.06 0.175 C 0.08 0.08 0.25 1.50 -0.04 1.50 -0.04 -0.04 -0.04 0.08 -0.04 0.08 0.175 G -0.04 0.08 0.25 -0.04 -0.04 -0.04 1.50 -0.04 0.68 0.45 0.68 0.08 0.325 T 0.19 -0.12 -0.08 -0.08 -0.08 -0.08 -0.08 0.97 0.05 -0.06 -0.06 -0.06 1.000 Sum 0.11 0.09 0.36 1.29 0.80 1.29 1.29 0.80 0.61 0.39 0.47 0.04 ! Imatrix = Ii, j i=1 A " j=1 w " A alphabet size (=4) ni,j, occurrences of residue i at position j w matrix width (=12) pi prior residue probability for residue i fi,j relative frequency of residue i at position j k pseudo weight (arbitrary, 1 in this case) f'i,j corrected frequency of residue i at position j Wi,j weight of residue i at position j Ii,j information of residue i at position j ! fi, j ' = ni, j + pik ni, j i=1 A " + k ! Ii, j = fi, j ' ln fi, j ' pi " # $ % & ' Reference: Hertz (1999). Bioinformatics 15:563-577. ! Ij = Ii, j i=1 A "
- 14. 14 Information content Iij of a cell of the matrix ? For a given cell of the matrix ? Iij is positive when fˇŻij > pi (i.e. when residue i is more frequent at position j than expected by chance) ? Iij is negative when fˇŻij < pi ? Iij tends towards 0 when fˇŻij -> 0 (because limitx->0 x*ln(x) = 0)
- 15. 15 Information content of a column of the matrix ? For a given column i of the matrix ? The information of the column (Ij) is the sum of information of its cells. ? Ij is always positive ? Ij is always positive ? Ij is 0 when the frequency of all residues equal their prior probability (fij=pi) ? Ij is maximal when ? the residue im with the lowest prior probability has a frequency of 1 (all other residues have a frequency of 0) ? and the pseudo-weight is 0 ! Ij = Ii, j i=1 A " = fi, j ' ln fi, j ' pi # $ % & ' ( i=1 A " ! im = argmini (pi ) k = 0 max(Ij )=1*ln( 1 pi ) = "ln(pi )
- 16. 16 ! Imatrix = Ii, j i=1 A " j=1 w " ! P site( ) " e#Imatrix Information content of the matrix ? The total information content represents the capability of the matrix to make the distinction between a binding site (represented by the matrix) and the background model. ? The information content also allows to estimate an upper limit for the expected frequency of the binding sites in random sequences. ? The pattern discovery program consensus (developed by Jerry Hertz) optimises the information content in order to detect over-represented motifs. ? Note that this is not the case of all pattern discovery programs: the gibbs sampler algorithm optimizes a log-likelihood. Reference: Hertz (1999). Bioinformatics 15:563-577.
- 17. 17 Information content: effect of prior probabilities ? The upper bound of Ij increases when pi decreases ? Ij -> Inf when pi -> 0 ? The information content, as defined by Gerald Hertz, has thus no upper bound.
- 18. 18 References - PSSM information content ? Papers by Tom Schneider ? Schneider, T.D., G.D. Stormo, L. Gold, and A. Ehrenfeucht. 1986. Information content of binding sites on nucleotide sequences. J Mol Biol 188: 415-431. ? Schneider, T.D. and R.M. Stephens. 1990. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res 18: 6097-6100. ? Tom SchneiderˇŻs publications online ? http://www.lecb.ncifcrf.gov/~toms/paper/index.html ? Papers by Gerald Hertz ? Hertz, G.Z. and G.D. Stormo. 1999. Identifying DNA and protein patterns with statistically significant alignments of multiple sequences. Bioinformatics 15: 563-577.