1 product development
•Download as PPTX, PDF•
1 like•1,230 views

This document provides an overview of product development from a regulatory perspective. It discusses the steps required to prove a product is safe and effective, including preclinical studies, investigational new drug application, clinical trials in phases 1-3, and the marketing application. The target product profile and good practices for manufacturing, clinical research, and laboratories are also covered. The goal is to help maintain high ethical standards and show favorable risk-benefit analysis for approval.


























![GXPs
• Good [anything] practices
• The common ones (these you have to know):
• GLP - Good Laboratory Practices
• GCP - Good Clinical Practices
• GMP - Good Manufacturing Practices
• GDP - Good Documentation Practices (more common in the US)
• Lesser used GXP
• GDP – Good Distribution Practices (more common in Europe)
• GRP - Good Review Practices (used by FDA)
• GCLP – Good Clinical Laboratory Practices
• GPP – Good Programming Practices](https://image.slidesharecdn.com/1productdevelopment-141209195430-conversion-gate02/85/1-product-development-30-320.jpg)










1 product development
- 1. Product Development Overview (Regulatory Perspective) Michael Matthews, RAC
- 2. Product Development Overview • Prove the product is safe and effective for the proposed indication • Pharmaceutical products are developed to help treat patients. Always remember that the patients are people and product development must maintain a very high ethical standard. • Risk/benefit • You also have to remember… • ~$1 - $5 billion taking into account failed products • Highly variable • Drugs for large populations cost more than drugs for rare diseases • Novelty • Hurry up and fail
- 4. Discovery to marketing http://www.phrma.org/innovation/clinical-trials
- 5. An API before and after development 10 years, $1 bil later An API into preclinical studies API after obtaining marketing approval Products do not change during the course of development, you are just learning more about it.
- 6. Target Product Profile (TPP) • The TPP is a document describing the goals and objectives of a product. • It includes a lot of information that will end up on the final label: • Indication and usage • Intended population • Dosage and administration • Dosage form/strengths • Acceptable adverse reactions • Expected or acceptable efficacy • Risk/benefit analyses • It is a living document and expected to be changed as testing progresses
- 7. TPP versus product label • FDA Draft Guidance: Target Product Profile http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatory Information/Guidances/ucm080593.pdf Keyruda Approved Label http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda _pi.pdf
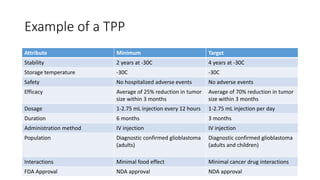
- 8. Example of a TPP Attribute Minimum Target Stability 2 years at -30C 4 years at -30C Storage temperature -30C -30C Safety No hospitalized adverse events No adverse events Efficacy Average of 25% reduction in tumor size within 3 months Average of 70% reduction in tumor size within 3 months Dosage 1-2.75 mL injection every 12 hours 1-2.75 mL injection per day Duration 6 months 3 months Administration method IV injection IV injection Population Diagnostic confirmed glioblastoma (adults) Diagnostic confirmed glioblastoma (adults and children) Interactions Minimal food effect Minimal cancer drug interactions FDA Approval NDA approval NDA approval
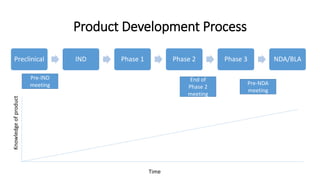
- 9. Preclinical IND Phase 1 Phase 2 Phase 3 NDA/BLA Time Pre-IND meeting End of Phase 2 meeting Pre-NDA meeting Product Development Process
- 10. Break
- 11. Preclinical studies (“IND enabling studies”) • In vitro assays • Genotoxicity • Drug interaction • In vivo studies • Pharmacokinetics • Toxicity (rodent and non-rodent) • Pharmacology and proof of concept • Immunogenicity • Other studies??? • More to come on this later *Research/discovery are generally not considered preclinical studies*
- 12. Pre-IND meeting Meeting with FDA to talk about: • Proposed indication • Proposed clinical plan • Summary information of the product • Proposed regulatory pathway • Summary of preclinical studies • Summary of manufacturing • Summary of the proposed clinical study(ies) • Other information Very strict meeting conduct Everything stated can and will be used against you.
- 13. Pre-IND meeting continued • Notify FDA that an IND is coming soon • Get agreement with FDA on certain regulatory concerns • Clinical study design • Preclinical data • Manufacturing methods • Other testing • Get FDA’s valuable comments
- 14. Investigational New Drug (IND) Application • Proposed indication • Example of the investigational product label • Summary of the preclinical and manufacturing information and any known clinical information (focus on clinical risk) • Complete manufacturing information • Where and how product is made • Controls used in the manufacturing process • Stability plan and known stability data • Other information • Complete preclinical reports, including all data • Full clinical protocol • Investigator qualifications • More to come on this later
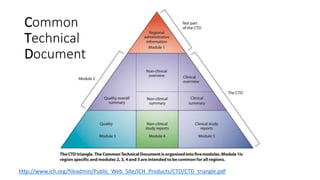
- 15. Common Technical Document http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/CTD/CTD_triangle.pdf
- 16. IND Amendments • Required IND amendments • IND Annual Reports • Safety Reporting and updated safety information • Meeting requests/general communications • Updated manufacturing information (stability data, mfg changes) • New clinical study protocols • New nonclinical study reports • New investigators/sites • Other IND amendments • Clinical study reports (CSRs) • IRB approvals • Informed consent forms • Updated investigators brochure or updated foreign market information • Nonclinical protocols
- 17. Nonclinical studies • Animal studies done in parallel with clinical studies • Pharmacokinetics • General toxicity • Chronic toxicity studies • Carcinogenicity • Food effect studies • Reproductive toxicity studies
- 18. Phase 1 clinical studies • 20-80 subjects • Purpose is usually pharmacokinetics or pharmacological effect • A lot of focus on safety • Usually open-label, healthy individuals • Exploratory and can be dose escalation • Usually a lot of analyses, a lot more visits • Product may not be completely “GMP” • GCP compliant
- 19. Phase 2 Clinical Studies • 200-400 subjects • Usually controlled • Focus on safety • Dose selection or dose determination • Can compare two investigational products to each other • Typically done in target population • GCP compliant
- 20. Phase 3 Clinical Studies • 1000+ subjects • Expected to be controlled and primary focus on efficacy • Evaluate risk-benefit analysis • These studies are to be close to the clinical use of the product • Usually at least 3 batches of product are used to show manufacturing consistency • Need 2 phase 3 studies for marketing approval • GCP compliant
- 21. Marketing application (NDA or BLA) 1. A lot of administrative information • Patenting information, labels and labeling, all of the communications with FDA • Postmarketing commitments and/or REMS 2. Summaries of preclinical, clinical, and manufacturing 3. Extensive manufacturing information 4. Complete preclinical and nonclinical study report (complete data) 5. Complete clinical reports (complete data and a lot of other information)
- 22. Expedited programs for serious conditions Fast Track Breakthrough Accelerated Priority Designation Designation Approval pathway Designation 505(b), FDAMA, FDASIA 506(a), FDASIA CFR314, 601, 506(c) PDUFA Serious + nonclinical data Serious + preliminary Serous + better than or qualified infectious clinical evidence + better available therapy + good disease product than available therapy surrogate endpoint Serious + significant improvement in safety or efficacy or supplement label change or QIPD or voucher Between IND – pre-NDA Between IND – EOP2 Before NDA With NDA or supplement 60 day response 60 day response N/A 60 day response Expedited review and Expedited review and Shorter review clock (6- rolling review rolling review months) http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM358301.pdf
- 23. Marketing Application Supplements • Annual Reports • Phase 4 or post-marketing studies • Submit reports to FDA on progress of the studies • Submit final reports once studies are completed • Most biopharmaceuticals require post-marketing studies • Safety reporting
- 24. Post-marketing changes • A change in indication most often requires clinical studies to prove efficacy • A change in manufacturing: • If extensive change – pre-approval needed • If moderate change – notify FDA within 30 days • If minor change – notify FDA in an annual report • Labeling/promotional changes • Online ads • TV commercials
- 25. Questions???
- 26. Break time!
- 27. Regulations and Guidance Documents • FD&C Act and other applicable laws: • http://www.fda.gov/RegulatoryInformation/Legislation/FederalFoodDrugand CosmeticActFDCAct/default.htm • All regulations are publically available – FDA’s interpretation of law • http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/cfrsearch.cfm • All guidance (FDA +ICH) publically available – FDA’s current thinking • http://www.fda.gov/Drugs/GuidancecomplianceRegulatoryInformation/Guida nces/default.htm
- 28. Applicable Regulations for Development • 21CFR11 – Electronic Records • 21CFR50 – Human Subject Protections • 21CFR54 – Financial Disclosure by Clinical Investigators • 21CFR56 – Investigational Review Boards • 21CFR58 – Good Laboratory Practices • 21CFR211 and other 200s – Good Manufacturing Practices, labeling, ads • 21CFR312 – Investigational New Drug Regulations • 21CFR314 – New Drug Application • 21CFR316 – Orphan Drugs • 21CFR600s – Biologics
- 29. Applicable Regulations for Development • 21CFR25 – Environmental Impact Considerations • 21CFR200s – Good Manufacturing Practices, labeling, ads • 21CFR312 – Investigational New Drug Regulations • 21CFR314 – New Drug Application • 21CFR316 – Orphan Drugs • 21CFR600s – Biologics • 45CFR46 – HHS Policy for Protection of Human Research Subjects
- 30. GXPs • Good [anything] practices • The common ones (these you have to know): • GLP - Good Laboratory Practices • GCP - Good Clinical Practices • GMP - Good Manufacturing Practices • GDP - Good Documentation Practices (more common in the US) • Lesser used GXP • GDP – Good Distribution Practices (more common in Europe) • GRP - Good Review Practices (used by FDA) • GCLP – Good Clinical Laboratory Practices • GPP – Good Programming Practices
- 31. Good Manufacturing Practices (GMP) • GMPs are meant primarily for the manufacturing process, testing, packaging, and labeling of pharmaceutical substances and products. • GMPs are defined in the regulations under 21 CFR 211 for finished pharmaceuticals. Ensure products are manufactured as to be safe and effective. • Helps prevent products from getting contaminated and helps to ensure products do what they are supposed to do.
- 32. Examples of FDA GMP Guidance Documents • ICH Q7A – Good Manufacturing Practice Guidance for API • ICH Q1A – Stability Testing of New Drug Substances and Products • ICH Q2B – Validation of Analytical Procedures: Methodology • cGMP for Phase 1 Investigational Drugs • Comparability Protocols – Protein Drug Products and Biological Products • Process Validation: General Principles and Practices • Q&A on cGMP for Drugs • Quality System Approach to Pharmaceutical cGMP Regulations http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm064971.htm
- 33. Good Clinical Practices (GCP) • Good Clinical Practices (GCP) are meant primarily for the conduct of clinical studies using any pharmaceutical products. • GCP principles are stated in the laws and regulations which govern clinical studies. All clinical studies must comply with these GCPs. • An international organization known as ICH published the most popular and most used GCP guidance (ICH E6) • Ensures clinical studies are conducted ethically and human subjects are protected • Ensures the scientific integrity of the study http://www.fda.gov/scienceresearch/specialtopics/runningclinicaltrials/default.htm
- 34. Examples of FDA GCP Guidance Documents • ICH E6 – Good Clinical Practice • ICH E3 – Structure and Content of Clinical Study Reports • ICH E9 – Statistical Principles for Clinical Trials • Adaptive Design Clinical Trials for Drugs and Biologics • Alzheimer’s Disease: Developing Drugs for the Treatment of Early Stage Disease • Standards for Clinical Trial Imaging Endpoints
- 35. Good Laboratory Practices (GLP) • Good Laboratory Practices are mentioned by name in the regulations (21 CFR 58) • Meant primarily for animal (in vivo) studies, but may also be referenced for the isolated cell (in vitro) studies • Ensures quality study design and the study is conducted in an ethical and appropriate matter • Ensure scientific integrity of the data collected
- 36. Examples of FDA Preclinical Development Guidance Documents • Good Laboratory Practices Q&A • ICH S1A – The Need for Long-term Rodent Carcinogenicity Studies • ICH S2 – Genotoxicity • ICH S3 – Toxicokinetics/pharmacokinetics • ICH S4 – Chronic Toxicity • Product Development Under the Animal Rule • Immunogenicity Evaluation of INDs • Nonclinical Safety Evaluation of Drug or Biologic Combinations • Safety Testing of Drug Metabolites
- 37. Good Documentation Practices (GDP) • GDPs is an industry term used to define how documentation should be done in a regulated environment • Applies to all disciplines: manufacturing, preclinical, and clinical • Ensures all information is captured accurately and all changes are captured and traced. • Mostly applies to handwritten documents (batch records, handwritten medical records, or handwritten results from assays). • For electronic documentation, a different term is used…Part 11 • ALCOA (attributable, legible, contemporaneous, original and accurate)
- 38. Other Examples Useful Guidance Documents • Part 11, Electronic Records; Electronic Signatures – Scope and Application • ICH M(series): Electronic Common Technical Document (submission standards) • Providing Submissions in Electronic Format – Standardized Study Data • Format and Content of Proposed Risk Evaluation and Mitigation Strategy (REMS) • Formal Meetings Between the FDA and Sponsor • Pharmacogenomic Data Submissions • Target Product Profile
- 39. More Resources • Manual of Policies and Procedures (MAPP) • http://www.fda.gov/aboutfda/centersoffices/officeofmedicalproductsandtob acco/cder/manualofpoliciesprocedures/default.htm • Staff Manual Guides (SMG) • http://www.fda.gov/AboutFDA/ReportsManualsForms/StaffManualGuides/de fault.htm • Data standards • http://www.fda.gov/ForIndustry/DataStandards/ • Orphan Drug Designations and Approvals • http://www.accessdata.fda.gov/scripts/opdlisting/oopd/ • Standards Organizations • USP, CDISC, OECD, HL7, CMS, ICD-9CM(10CM)
- 40. And More Resources (specific products) • Warning Letters • http://www.fda.gov/iceci/enforcementactions/WarningLetters/default.htm • Clinical Investigator Inspection Search/BIMO • http://www.accessdata.fda.gov/scripts/cder/cliil/index.cfm • http://www.accessdata.fda.gov/scripts/cder/bmis/index.cfm • Drugs@FDA • http://www.accessdata.fda.gov/scripts/cder/drugsatfda/ • Public Meetings and Advisory Committee Meetings • http://www.fda.gov/AdvisoryCommittees/CommitteesMeetingMaterials/default.htm • ClinicalTrials.gov • www.clinicaltrials.gov • Patents • http://patft.uspto.gov/