AB-RNA-alignments-2010
âĒ
0 likesâĒ186 views

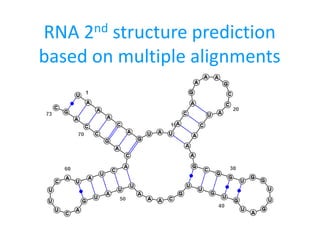
RNA secondary structure can be predicted using comparative sequence analysis of multiple RNA alignments. Conserved base pairs are often revealed by frequent compensatory mutations that maintain base pairing complementarity. The covariance method measures sequence covariation between columns in an alignment using mutual information to identify compensatory mutations. Predictions start with an initial alignment that is then refined based on the predicted secondary structure in an iterative process. Stochastic context-free grammars can be modified to generate RNA secondary structure by modeling the probability of columns being single or forming base pairs.









AB-RNA-alignments-2010
- 1. RNA 2nd structure prediction based on multiple alignments
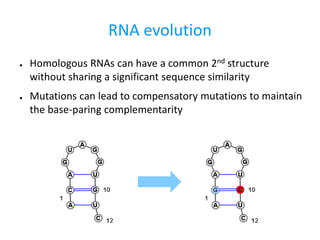
- 2. RNA evolution â Homologous RNAs can have a common 2nd structure without sharing a significant sequence similarity â Mutations can lead to compensatory mutations to maintain the base-paring complementarity
- 3. Comparative sequence analysis â In a structurally correct multiple alignment of RNAs, conserved base pairs are often revealed by the presence of frequent correlated compensatory mutations â Measure sequence covariation: mutual information â is the frequency of one of the four bases observed in col I â is the joint frequency of the base pairs observed in columns i and j ððð = â ðĨ ð,ðĨ ð ððĨ ð,ðĨ ð log2 ððĨ ð,ðĨ ð ððĨ ð â ððĨ ð ððĨ ð ððĨ ð,ðĨ ð
- 4. Covariance method G U C U U C G G A C G A C U U C G G U C G G C U U C G G C C ð2,9 = 3 â 1 3 â log2 1/3 1/9 = log23 â 1.59 ððð = â ðĨ ð,ðĨ ð ððĨ ð,ðĨ ð log2 ððĨ ð,ðĨ ð ððĨ ð â ððĨ ð â Mij varies between 0 and 2 â Mij is 2 when i and j appear completely random but are perfectly correlated â if i and j are uncorrelated, the mutual information is 0 â if either i or j are highly conserved positions, we also get little or no mutual information
- 5. â Mij is 2 when i and j appear completely random but are perfectly correlated â if i and j are uncorrelated, the mutual information is 0 â if either i or j are highly conserved positions, we also get little or no mutual information Covariance method G U C U U C G G A C G A C U U C G G U C G G C U U C G G C C G C C U U C G G G C ð1,9 = 4 â 1 4 â log2 1/4 1/4 = 0 ððð = â ðĨ ð,ðĨ ð ððĨ ð,ðĨ ð log2 ððĨ ð,ðĨ ð ððĨ ð â ððĨ ð ð2,9 = 4 â 1 4 â log2 1/4 1/16 = 2
- 6. Comparative analysis â Start with a multiple alignment â Predict 2nd structure base on alignment â Refine alignment based on 2nd structure â Repeat â The sequences to be compared must be sufficiently: â similar that they can be initially aligned by primary sequence â dissimilar that a number of covarying substitutions can be detected
- 7. Comparative analysis â How to build 2nd structure based on alignment? â Greedy method â choose the pair of columns that have the highest Mij â make a base pairs â carry on with the second highest Mij â problem columns might end up in more than one base pair
- 8. SCFGs and RNA alignments â An SCFG could be modified to generate columns of alignments instead of nucleotides â Requires a fixed number of sequences in the alignment â Instead, change it to generate the structure! ð â . ð âĢ ð. ð ðð Îĩ ð â ðð âĢ ðð âĢ ðð âĢ ðĒð ðð âĢ ðð âĢ ðð âĢ ððĒ ðððĒ âĢ ððð âĢ ðððĒ ðĒðð âĢ ððð âĢ ðĒðð ðð Îĩ
- 9. SCFGs and RNA alignments â How to determine the probability of a structure for a given sequence? â A C G U C G U C â ( ( ( . ) ) ) . â Use CYK to calculate the maximum probability of a structure for a given sequence... ð â ð. â ð . â ð . â ð . â . ð . â . .
- 10. SCFGs and RNA alignments â Use a phylogenetic tree (including branch lengths) to: â determine the probability of a column to be single â determine the probability of two columns to form a base pair â Use the SCFG and the columns probability to determine the best secondary structure for the alignment â CYK and the other SCFGs algorithms are basically the same
- 11. SCFGs and RNA alignments Knudsen&Hein 1999