






































More Related Content
What's hot (20)
Similar to Calibration (20)


















Recently uploaded (20)




















Calibration
- 1. CALIBRATION Ms.R.L. Mhetre Asst. Professor Modern College of Pharmacy, Moshi, Pune
- 2. CALIBRATION: It is comparison between the reference standard and instrument to be calibrated. Reference standard material used for calibration should be calibrated according to ISO/IEC 17025. GOAL: • TO MINIMIZE ANY MEASUREMENT UNCERTAINITY BY ENSURING THE ACCURACY OF THE TEST EQUIPMENT. • CALIBRATION QUALIFIES AND CONTROLS ERRORS OR UNCERTAINITIES WITHIN MEASUREMENT PROCESS TO AN ACCEPTABLE LEVEL. • TO DETERMINE WHETHER EQUIPMENT IS STILL FIT FOR ITS INTENDED USE. Where to be calibrate • I/II/III PARTY LABORATORY • ACCREDITED CALIBRATION LABPRATORY- MANUFACTURER/SUPPLIER LABORATORY.
- 3. BASIC REQUIRMENT OF CALIBRATION • Location: Permanent • Environment: temp, humidity, airflow, filtration, noise level, vibrations, cleanliness, power supply, calibartion areas • Equipment: Reference standard, working standard, accessories, computer system, software • Staff: training, responsibility, technical competence, authority • Management system: document, calibration record.
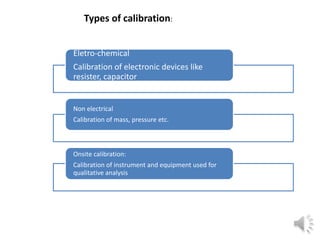
- 4. Types of calibration: Eletro-chemical Calibration of electronic devices like resister, capacitor Non electrical Calibration of mass, pressure etc. Onsite calibration: Calibration of instrument and equipment used for qualitative analysis
- 5. Purpose/importance of calibration: • To maintain the accuracy and precision of test equipment at all times. • It is used to detect, correlate, report or eliminate any variation in the accuracy of the equipment being calibrated. • Component and equipment can degrade due to changes in temperature/humidity or sustain mechanical stress. This is called drift. Then test results become unreliable. Drift can not be eliminated, it can be detected and either corrected or compensated by the process of calibration. • Properly calibrated equipment provides confidence that your product/ services/results meets their specifications. • Calibration optimizes resources, ensure consistency, ensures measurements are compatible with those made elsewhere.
- 6. Calibration frequency It is depending on following points: • Classification of critical or non-critical • Usage (light/heavy usage) • Handling (light/heavy handling) • Manufacturers recommendations • Reference to accreditation body guidelines for a specific measurement system. Basic criteria for establishing the frequency of calibration shall consider following conditions: • Manufacturers recommendations • Standard and relevant procedures • Instrument past behavior • Overall impact of non-compliance in the calibration process • Previous experience of laboratory technical staff. • Critical assessment team shall be responsible for the definition and approval of the frequency of calibration. The critical process measuring instruments shall be calibrated at least twice a years.
- 7. Methods to determine calibration frequency: • Quantitative • Qualitative • Quantitative: In this method, the initial frequency of calibration is determined by the product of the three impact factors described by equation: OF = WF × FF × LF OF ----OVERALL FACTOR WF---WEAR FACTOR FF----FREQUENCY OF USE LF---- LOCALIZATION CONDITION FACTOR
- 8. WF FF LF High= 9 or 10 High= 9 or 10 Every day= 9 or 10 Moderate = 6, 7 or 8 Moderate = 6, 7 or 8 Every week = 6, 7 or 8 Low = 3,4 or 5 Low = 3,4 or 5 Every month= 3, 4 or 5 Very low= 1 or 2 Very low= 1 or 2 Every year or twice a year= 1 or 2
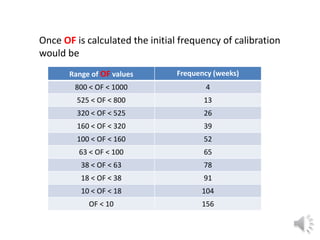
- 9. Once OF is calculated the initial frequency of calibration would be Range of OF values Frequency (weeks) 800 < OF < 1000 4 525 < OF < 800 13 320 < OF < 525 26 160 < OF < 320 39 100 < OF < 160 52 63 < OF < 100 65 38 < OF < 63 78 18 < OF < 38 91 10 < OF < 18 104 OF < 10 156
- 10. QUALITATIVE: Here, the initial frequency of calibration is selected according to the following points: 1. Characteristics: Degree of importance of the variable to be controlled to ensure the quality of product to be manufactured and it can be designed as critical, significant, important or not special. 2. Intensity of use: Rare, often and quiet often. 3. Environment: Loss of calibration due to different operating conditions is a critical factor to be considered in the definition of the frequency of calibration. These factors can be classifies as under control, moderate and aggressive.
- 11. Thank you