



































Cystic diseases of kidney
- 1. CYSTIC DISEASES OF KIDNEY Dr Anshita Dubey
- 5. Bonsib (2009) Classification of Renal Cystic Diseases and Congenital Anomalies of the Kidney and Urinary Tract I. Polycystic renal diseases • Autosomal-recessive polycystic kidney disease • Autosomal-dominant polycystic kidney disease • Acquired renal cystic disease • Glomerulocystic kidney diseases III. Tubulointerstitial syndromes +/- cysts A. Renal tubular dysgenesis B. Nephronophthisis C. Medullary cystic diseases D. Bardet-Biedel syndromes
- 6. IV. Cystic neoplasms and neoplastic cysts • Cystic nephroma • Mixed epithelial and stromal tumor • Multilocular cystic renal cell carcinoma • Tubulocystic renal cell carcinoma • Von Hippel–Lindau disease • Lymphangioma/hygroma renalis • Cystic partially differentiated nephroblastoma V. Miscellaneous cysts • Simple cortical cysts • Medullary sponge kidney • Localized renal cystic disease
- 7. PATHOGENESIS
- 10. Autosomal recessive polycystic kidney disease • mutations of a gene on chromosome 6p named polycystic kidney hepatic disease gene (PKHD1) • related protein polyductin (fibrocystin) - epithelial cells of the collecting ducts, thick ascending loops of Henle, biliary and pancreatic duct epithelia
- 11. • neonatal period - renal symptoms • ‘Potter’ phenotype • POTTER’S SEQUENCE RENAL FAILURE OLIGOHYDRAMINIOS COMPRESSION AND STRUCTURAL ABNORMALITIES PULMONARY HYPOPLASIA • perinatal period – renal failure and portal hypertension
- 12. • Later in life - congenital hepatic fibrosis with a biliary dysgenesis and bile duct ectasia • Hepatic portal hypertension with hepatosplenomegaly and esophageal varices Gross-The cysts tend to be linear and radiate from the medulla to the outer cortex. Develop in the collecting ducts, which expand to a large size due to fluid accumulation within the cyst cavity. Microscopically- the cysts appear as dilated tubular structures lined by cuboidal or flattened epithelium
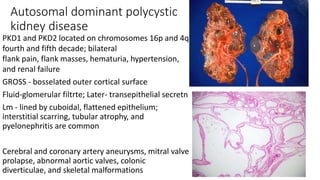
- 13. Autosomal dominant polycystic kidney disease PKD1 and PKD2 located on chromosomes 16p and 4q fourth and fifth decade; bilateral flank pain, flank masses, hematuria, hypertension, and renal failure GROSS - bosselated outer cortical surface Fluid-glomerular filtrte; Later- transepithelial secretn Lm - lined by cuboidal, flattened epithelium; interstitial scarring, tubular atrophy, and pyelonephritis are common Cerebral and coronary artery aneurysms, mitral valve prolapse, abnormal aortic valves, colonic diverticulae, and skeletal malformations
- 14. Acquired renal cystic disease • Three or more cysts per kidney in patients on longstanding hemo- or peritoneal dialysis for end stage renal disease Pathophysiology - May be due to uremia due to obstruction by oxalate crystals, fibrosis or hyperplasia Micro : Cysts lined by flattened or cuboidal epithelium that may show focal pseudopapillae with nuclear enlargement and loss of polarity • Cysts may contain oxalate crystals • Surrounding parenchyma shows global glomerulosclerosis, interstitial fibrosis & tubular atrophy.
- 15. Glomerulocystic kidney disease • common in newborns and young children • associated with ADPKD genes • BERNSTEIN’S CLASSIFICATION
- 16. • dilatation of Bowman’s space in the plane of section of two- to threefold that of normal
- 18. Nephronophthisis • autosomal recessive • most frequent genetic cause of ESRD in children and adolescents • six genes (NPHP1, NPHP2/inversin, NPHP3, NPHP4, NPHP5, and NPHP6) - nephrocystins, which are expressed in primary cilia, basal bodies, or centrosomes • inability to concentrate urine, polydipsia, aneuresis, severe anemia, and growth retardation • Bilateral • reduced in size ; granular surface. • On c/s, the cortex and medulla are both thinned. The corticomedullary junction – indistinct; thin-walled, fluid-filled cysts
- 19. • Tubulointerstitial fibrosis with lymphocytic inflammatory infiltrate, tubular atrophy, and cyst formation; marked thickening of the tubular basement membrane (PAS-stained sections)
- 20. Medullary cystic kidney disease • rare autosomal dominant disorder • MCKD1 and MCKD2 genes chromosomes 1q and 16p • third and fourth decades of life ̴juvenile NPH (-growth retardation and long history of anemia) normal or moderately reduced in size with small corticomedullary cysts. • Microscopically, diffuse interstitial inflammation with fibrosis and tubular atrophy interspersed with hypertrophied and dilated tubules
- 21. Renal tubular dysgenesis. • poor development of proximal tubules, early onset and persistent anuria -oligohydramnios and the Potter sequence Inherited RTD is genetically heterogeneous and linked to mutations in the genes encoding the major components of the renin-angiotensin system (RAS)
- 22. Secondary RTD • Twin-to-twin transfusion syndrome • Foetal exposure to RAS blockers
- 23. Bardet-Biedl syndrome • Primary Features -Rod-cone dystrophy. Postaxial polydactyly. Truncal obesity Learning disabilities. Hypogonadism (males)/genital abnormalities (females) Renal anomalies. • Secondary Features -Speech delay/disorder. Developmental delay. Behavioral abnormalities. Eye abnormalities Brachydactyly/syndactyly. Diabetes mellitus. Orodental abnormalities; craniofacial dysmorphism Cardiovascular anomalies, Hepatic involvement Hirschsprung disease Anosmia.
- 24. Cystic neoplasms and neoplastic cysts
- 25. Cystic nephroma • benign cystic lesion of the kidney of unknown etiology • before age 2 and after age 40. (before 2- males, after 40 – females) • conglomeration of noncommunicating cysts of varying size; pseudocapsule around cysts- compression of adjacent renal parenchyma • lined with flattened, cuboidal cells with hobnail-appearing cells
- 26. • DIAGNOSTIC CRITERIA : (a) lesion must be multilocular (b) the cyst for the most part lined by epithelium (c) the cyst must not communicate with renal pelvis (d) the residual renal tissue should be essentially normal, except for pressure atrophy (e) no fully developed nephrons. IHC • cytokeratin 19, cytokeratin AE1/AE3, epithelial membrane antigen • ovarian-like stroma stain positively for progesterone receptors and estrogen receptors
- 27. Mixed epithelial and stromal tumor • solid and cystic, tan to yellow Well circumscribed but unencapsulated tumor in renal pelvis • immunoreactivity for desmin and smooth muscle actin • MEST – higher stromal to epithelial ratio prominent ovarian stroma smaller cysts with phyllodes glands pattern stromal luteinisation CN - large cysts, thin septa, and low stromal to epithelial ratio
- 28. Multilocular cystic renal cell carcinoma •mean age of 51 years •excellent outcome •Gross – encapsulated non communicating multiloculated lobules filled with gelatinous fluid. •Micro - lining of cysts may show multilayering, cells with granular to clear cytoplasm No expansile growth of clear tumor cells / solid nodules •Cystic RCC - cystic degeneration of conventional RCC (worse prognosis)
- 29. • CD10, vimentin, and epithelial membrane antigen (EMA).
- 30. Tubulocystic renal cell carcinoma • mean age of 60 years male preponderance • Gross - circumscribed, unencapsulated, and cortical in location. “bubble-wrap” or “Swiss cheese” appearance • Micro - lined by a single layer of neoplastic cells which range from flat, hobnail, cuboidal, cylindrical to columnar. They are separated by thin, hypocellular fibrous septa that lack ovarian-type stroma or desmoplasia,
- 31. • Immunohistochemically, positive - markers of PCT (CD10, carbonic anhydrase IX, vimentin, AMACR) as well as distal tubule/collecting duct (CK7, CK19, parvalbumin, BerEp4).
- 32. Von Hippel–Lindau disease • AD • 3p • Multiple cysts in the kidneys, pancreas and genital tract • Hemangioblastomas • clear cell renal cell carcinoma, pheochromocytoma, pancreatic neuroendocrine tumor, epididymal cystadenomas and endolymphatic sac tumors
- 33. Lymphangioma/hygroma renalis • Extermely rare type of lymphangioma located in the pericalyceal area • multiloculated cyst extending to the pericalyceal and hilar area covering the ureter • multicystic lesion with flat endothelial cells • cytokeratin and calretinin negative • presence of endothelial lined lymphatic channels separated by the connective tissue is the main histologic feature of the disease.
- 35. SIMPLE RENAL CYST • most common • occurrence increases with age • Asymptomatic • translucent, filled with clear serous fluid and lined by a single layer of cuboidal or flattened epithelium. • complicated – thick capsular walls with hemosiderin-laden macrophages and atrophic lining epithelium
- 36. Medullary sponge kidney • tubular ectasia of the collecting ducts and cystic formation confined to the medullary pyramids • cysts are multiple, small, and limited to the medullary pyramids and papillae • The cysts are lined by collecting duct epithelium and usually communicate with collecting tubules • The interstitium often shows severe inflammation and scaring, frequently accompanied by tubular atrophy
- 37. THANK YOU