Cysticfibrosis
âĒDownload as PPT, PDFâĒ
3 likesâĒ1,286 views

Cystic fibrosis is a genetic disorder caused by a mutation in the CFTR gene located on chromosome 7. This gene encodes for the CFTR protein, which functions as a chloride channel in epithelial cells. The most common mutation is a deletion of three nucleotides coding for phenylalanine at position 508 (ÎF508), which causes the CFTR protein to fold incorrectly in the cell. As a result, it does not traffic to the cell membrane properly and cannot regulate chloride transport. This leads to a buildup of thick mucus in the lungs, pancreas and other organs, causing chronic issues like lung infections and problems with digestion. The disease primarily affects the white population and results from inheriting two copies of the mutated gene















Cysticfibrosis
- 1. CYSTIC FIBROSIS MOLECULAR BASIS OF THE DISEASE & CHANNELS INVOLVED COURSE NO. 629
- 2. INTRODUCTION ïķCystic fibrosis is a chronic, progressive, life threatening genetic disorder of pediatrics. ïķIt affect white population (1 in 3200 live births) but is uncommon among Asian and African population. ïķIt affects exocrine glands (mainly sweat glands) and mucus gland present on the epithelial lining of lungs, pancreas, intestine, and reproductive system. ïķCF is a defect in epithelial chloride channel protein, causes membrane to become impermeable to Chloride ion.
- 3. GENETICS OF THE DISEASE: ïąCF is autosomal recessive disease means: ïąChildren who inherit two abnormal alleles -one from each parent- will have CF ïąChildren who inherit only one mutant gene will be carrier of the disease
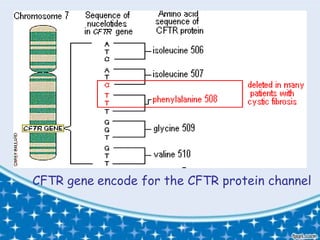
- 4. CF occurs due to the deletion of 3 nucleotides which code for the phenylalanine from the CFTR (cystic fibrosis transmembrane conductance regulator) gene located on chromosome no.7 at position 508 This mutation is known as ÎF 508 ITâS IN THE GENES!
- 5. CFTR gene encode for the CFTR protein channel
- 6. STRUCTURE OF THE CFTR PROTEIN: ïžCFTR protein is a cAMP induced Channel made up of five domains: ïžTwo membrane-spanning domain (MSD1 & MSD2) that form ClÂŊ ion channel. ïžTwo nucleotide binding domains (NBD1 & NBD2) that bind and hydrolyze ATP. ïžA regulatory R domain.
- 7. REGULATION OF CFTR PROTEINREGULATION OF CFTR PROTEIN Several proteins bind with CFTR and regulate itâs activity. Protein phosphatase 2A (PP2A), AMP kinase (AMPK), syntaxin-1A (SYN1A), synaptosome associated protein 23kD (SNAP23) ,mammalian uncoordinated 18a (Munc-18a) inhibit channel activity and channel mediated transport Na/H exchanger regulatory factor isoform-1 (NHERF-1), receptor for activated C-kinase (RACK1), protein kinase C (PKC), protein kinase A (PKA) and ezrin, radixin, moesin binding domain (MBD), NBD, Phosphatidylinositol bisphosphate, Rho and regulatory domain enhance the activity of CFTR
- 8. ASSEMBLY OF PROTEINS WITH CFTR PROTEIN
- 9. ÎF508 mutations result in the defected NBD1 domain due to which protein is folded incorrectly. Recognized, marked and degraded by the cell quality control mechanism when reaches to the ER Protein does not reaches to the apical membrane of epithelium
- 10. PROGRESSION OF THE DISEASEPROGRESSION OF THE DISEASE In Respiratory epithelium, function of CFTR is to secrete chloride ion Loss of CFTR function causes Loss or reduction of Cl ion in luminal secretion Followed by active luminal Na absorption through ENaC Increases passive water absorption from the lumen. Impaired mucociliary action, accumulation of thick, viscous, dehydrated mucus
- 11. Obstruction of air passage and recurrent pulmonary infections
- 12. In pancreas, reduction in the luminal water content of the secretion, thick mucus, will cause clogging of protein and obstruction in ductules and acini of pancreas Due to which digestive enzymes of pancreas will not reach to the intestine. Same is the condition in Liver, along with insufficient secretion of bicarbonate ion. Thus bile is also not reached to the intestine. Digestive enzyme insufficiency will result poor absorption of proteins, fats and fat soluble vitamins such as vitamin A, D, E, and K. Gastrointestinal abnormalities secondary to disease and growth abnormalities.
- 13. In SWEAT GLANDS CFTR is responsible for reabsorbtion of Cl ion along with Na ion through epithelial Na channel (ENaC). Impaired function of CFTR cause the production of hypertonic salty sweat, and ultimately dehydration.
- 16. References: Robbins Basic Pathology, 9th edition. Exploring Genes and Genetic Disorders http://www.ornl.gov/sci/techresources/Human_Genome/posters/chromos The Encyclopedia of Science http://www.daviddarling.info/encyclopedia/ETEmain.html Nature reviews Molecular Cell Biology http://www.nature.com/nrm/index.html THANK YOU
Editor's Notes
- #2: MOLECULAR PHYSIOLOGY AND BIOPHYSICS OF ION CHANNELS COURSE NO.629
- #3: oteinINTRODUCTION OF THE DISEASE.
- #4: GENETICS OF THE DISEASE
- #5: GENETICS OF THE DISEASE
- #6: GENETICS OF THE DISEASE
- #7: MOLECULAR BASIS OF THE DISEASE
- #9: Several proteins bind with CFTR and regulate itâs activity. Protein phosphatase 2A (PP2A), AMP kinase (AMPK), syntaxin-1A (SYN1A), synaptosome associated protein 23kD (SNAP23) ,mammalian uncoordinated 18a (Munc-18a) inhibit channel activity and channel mediated transport Na/H exchanger regulatory factor isoform-1 (NHERF-1), receptor for activated C-kinase (RACK1), protein kinase C (PKC), protein kinase A (PKA) and ezrin, radixin, moesin binding domain (MBD), NBD, Phosphatidylinositol bisphosphate, Rho and regulatory domain enhance the activity of CFTR
- #10: Hypothetical structure of the protein, otherwise abnormally coiled CFTR protein does not reaches to the membrane.