
































































More Related Content
What's hot (20)
Similar to Handling OOS results (20)


















Handling OOS results
- 1. HANDLING OF OOS RESULTS Sasha Nezlin VP Analytical Laboratories Nextar chempharma solutions Ltd.
- 2. HISTORY … not so long ago … 1992 … 1993 … 1992 European Union is formed Yugoslavia disintegrates Microsoft ships Windows 3.1 Suicide bombing at Israel Embassy in Buenos Aires EuroDisney opens near Paris Yitzhak Rabin wins the elections Bill Clinton wins the elections United States of America, Plaintiff V. Barr Laboratories Inc., Defendants, Civil Suite 92-1744 August 17 till October 12, 1992 Judge Wolin decision on February 5, 1993 1993 (Jan-Feb) Václav Havel elected President of the Czech Republic World Trade Center bombing History … Very actual today HANDLING OF OOS RESULTS
- 3. HANDLING OF OOS RESULTS
- 4. USA – FDA CDER OOS GUIDANCE - 2006 EDITION HANDLING OF OOS RESULTS
- 5. CANADA - GMP GUIDELINES - 2009 EDITION HANDLING OF OOS RESULTS
- 6. PIC/S - GMP GUIDELINES - 2009 EDITION HANDLING OF OOS RESULTS
- 7. MHRA – GUIDELINE ON OOS RESULTS – COMING IN 2010? HANDLING OF OOS RESULTS
- 8. MHRA – GUIDELINE ON OOS RESULTS – COMING IN 2010? HANDLING OF OOS RESULTS
- 9. Q6A: Specifications : Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances DEFINITION OF SPECIFICATIONS HANDLING OF OOS RESULTS
- 10. DEFINITION OF ?RESULTS? OOS Results - Test results laying outside of the specifications Questionable results (e.g. close to spec / limit) Out of trend ( OOT - stability, but not only) Out of limits ( OOL - alert / alarm) Unexpected Results results that are aberrant, abnormal, anomalous, atypical, irregular or deviant Batch failure , used by FDA, disliked by Judge Wolin (Barr case) Additional unexpected peaks in chromatogram (e.g. Dissolution test) HANDLING OF OOS RESULTS
- 11. A History of the OOS Problem, Steven Kuwahara (BioPharm International, Volume 20, Issue 11) The most significant abuse of statistical methods has been to test lots repeatedly until a sample falls within the specification range, and then to accept a lot based on one passing result. This method is known as " testing into compliance “. In "testing into compliance," an unethical manufacturer hopes that even a bad lot will produce a passing test result, as a result of extreme statistical variation. Consequently, failing test results are ignored and retests are ordered until extreme variation produces a passing test result. The passing result is accepted and the lot is released based on that result. … seven to eight retests were ordered in an attempt to obtain a passing result ... There is no reason to believe that the results obtained from the retests are really different from the original test result . HANDLING OF OOS RESULTS
- 12. Investigations of OOS results should be performed in cases of : Batch release testing and testing of starting materials. IPC testing: if data is used for batch calculations/decisions and if in a dossier and on Certificates of Analysis. NOTE : OOS NOT applicable for in-process testing used for process adjustment (eg - pH, viscosity, etc.), and for process validation studies (variable parameters, analytics - robustness). Stability studies on marketed batches of finished products and or active pharmaceutical ingredients, ongoing / follow up stability ( NOT applicable for stress/forced degradation tests ). If the previous released GMP batch used as reference sample in an OOS investigation produced OOS or questionable results  investigation should be extended to include it. Batches for clinical trials  under cGMP’s. HANDLING OF OOS RESULTS
- 13. Sun Pharmaceutical Industries Inc 8/25/2010 During release testing, batches 90056 and 90057 of Promethazine Hydrochloride (HCI) Tablets, 25 mg, exhibited OOS water content results of 5.7% and 5.9%, respectively (the specification is …). The OOS results were invalidated after a retest yielded acceptable results, despite your failure to identify an assignable laboratory cause. Furthermore, you failed to extend the investigation to associated batches . The investigation did not include batch 90058 that was analyzed in conjunction with batches 90056 and 90057 and for which passing results were obtained. Yet your Quality Control Unit (QCU) released lots 90056A, 90057A, and 90058A between May 2009 and June 2009. FDA Warning letters, Aug-Sept-Oct 2010 HANDLING OF OOS RESULTS
- 14. Kyowa Hakko Kogyo Co., Ltd. 9/29/10 1. Failure of your quality control unit/laboratory to thoroughly investigate and document out-of-specification (OOS) results obtained. For example, … b) Your firm's OOS investigation relating to impurity levels for (b)(4), lot (b)(4) , concluded that the root cause was a laboratory error, but the investigation did not identify what specific laboratory error occurred. … The investigational checklist initially indicated that no problem was found with the analysis. The investigational checklist you currently use is insufficient to detect and evaluate instrument problems and standard/sample preparation errors. You authorized retesting of (b)(4), lot (b)(4), without identifying a possible root cause . Instead, a new sample preparation was used to retest the product, which was found within specification. You used the passing retest results to invalidate the original OOS results, with no laboratory error attributed in obtaining the original result . This retesting approach lacks scientific justification. HANDLING OF OOS RESULTS
- 15. Contract Pharmacal Corporation 10/14/10 Your response … is inadequate because it fails to include details on how your firm handles investigations, such as which errors or incidents would result in a thorough out-of-specification (OOS) investigation … d) Ibuprofen had OOS assay result of 110.3% on (b)(4) April 12, 2010, for the initial testing of (b)(4) (Ibuprofen), under lot 102005. The analyst performed repeat testing, in duplicate, on the same date, from the same sample. After that, both assay test results were found within specification. Your response states, "It was determined that each of the OOS results was due to a procedural error on the part of the analyst." However, your response does not include any information regarding which part of the Standard Operating Procedure (SOP) the analyst failed to follow and whether you extended the investigation for the assay testing to associated batches. You should report all test results that have not been invalidated, and these results should be considered in batch release decisions . Finally, we note your commitment to retrain analysts on a routine basis, but your response fails to include timeframes for completing training to address OOS results. HANDLING OF OOS RESULTS
- 16. Evaluation of OOS results should include looking for possible statistics : Similar / multiple OOS results for certain analytical methods? Similar / multiple OOS results for certain instruments? - Similar / multiple OOS results for certain products? - Similar / multiple OOS results for certain analysts? Investigation : - The investigation has to follow a pre-established investigation plan - The investigation has to be well documented / summarized - The investigation is the basis for a release decision - The investigation is the basis for CAPA HANDLING OF OOS RESULTS
- 17. NEVER start or continue a test if SST or calibration criteria are not met NEVER continue tests that you expect to be invalidated at a later time for an assignable cause NEVER complete analysis for the sole purpose of seeing what results can be obtained when obvious or very likely errors have occurred NEVER knowingly produce an OOS result QC / analytical / contract labs must be aware and informed of any process deviation that may have a negative effect on the potency or purity of the product and may cause OOS results! HANDLING OF OOS RESULTS
- 18. All solutions and reagents must be retained until all data has been second person verified as being within the defined acceptance criteria. Compendial tests that are statistic in nature and/or measure variability (Dissolution, Uniformity of Dosage Units, Sterility) - have several levels for additional analyses with specific acceptance criteria (e.g. dissolution levels S1, S2 & S3; uniformity of dosage units testing of 20 additional units). However the sample test criteria is usually the first level of testing and if a sample has to be tested to the next level - this should be investigated/documented prior to proceeding to next level - as it is not an “ expected ” result. HANDLING OF OOS RESULTS
- 19. The amount of the initial sample should be sufficient for: - the initial testing - Possible investigation - Confirmation of the OOS results - Retained sample A lack of sample material is not necessarily a suitable reason for re-sampling for the purposes of OOS investigation Contract laboratory ( usually ) is not involved in sampling HANDLING OF OOS RESULTS
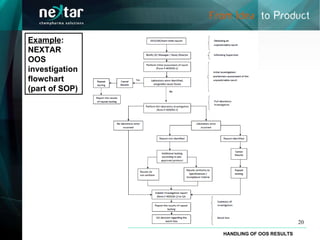
- 20. Example : NEXTAR OOS investigation flowchart (part of SOP) HANDLING OF OOS RESULTS
- 21. Sources of error - Correct test methodology followed (e.g. method version number) - Correct sample(s) tested - Sample integrity maintained, correct container and chain of custody - Assessment of the possibility that the sample contamination/degradation has occurred during the testing / re-testing procedure (e.g. sample left open to air) - All equipment used in the testing is within calibration date - Review equipment logbooks - Appropriate standards used in the analysis - Standards and/or controls performed as expected - System suitability conditions met (those before analysis and during analysis) - Correct and clean glassware used - Correct pipette / volumetric flasks volumes used - Media/Reagents prepared according to procedure - Items were within expiry date - A visual examination (solid and solution) - Correct specification applied - Data acceptance criteria met - The analyst is trained on the method - Interview analyst to assess knowledge of the correct procedure / performance - Examination of the raw data, including chromatograms and spectra - Any previous issues with this test / method / product - Other potentially interfering testing / activities occurring at the time of the test - Environmental conditions (temperature/humidity/draughts) during testing - Review of other data for other batches performed within the same analysis set - Consideration of other results (not only OOS) obtained for the tested batch - Review of method validation HANDLING OF OOS RESULTS
- 22. Example : NEXTAR OOS preliminary assessment checklist HANDLING OF OOS RESULTS
- 23. Example : NEXTAR OOS laboratory investigation form HANDLING OF OOS RESULTS
- 24. Example : NEXTAR OOS laboratory investigation form HANDLING OF OOS RESULTS
- 25. Initial laboratory assessment of the unexpected result Full laboratory Investigation Sampling Production Report / Decision 0 - 2 0 - 7 2 - 10 2 - 21 - 30 days Suggested timeline for the investigation (business days) HANDLING OF OOS RESULTS
- 26. Subcontractors – contract manufacturing and testing HANDLING OF OOS RESULTS
- 27. FDA OOS Guidance : For contract laboratories, the laboratory should convey its data, findings, and supporting documentation to the manufacturing firm’s quality control unit (QCU), who should then initiate the full-scale OOS investigation. Subcontractors – additional considerations Quality agreement – should relate to OOS investigations Should clearly define the expectations, timelines, agreed procedures, access to data, documentation, performing assessment, batch disposition – by contract giver! (if only testing performed by contract lab). OUT OF SPECIFICATION RESULTS HANDLING OF OOS RESULTS
- 28. DOCUMENT YOUR OBSERVATIONS IN REAL TIME! TIP : visual observation is an important part of testing and/or investigation of unexpected results. Therefore analysts (especially at contract lab!) may document the occurrences by taking photos – helping to document / verify observation of unexpected result – e.g. appearance of the sample, lack of disintegration or dissolution, etc. OUT OF SPECIFICATION RESULTS HANDLING OF OOS RESULTS
- 29. Costs of OOS investigation Preliminary assessment Analyst + Supervisor Documentation ! Full laboratory investigation Best case: 1 day, 8 hours Batch delay Documentation ! ! Deviation investigation QA investigation Other departments (Maintenance, Production, Packaging, etc.) Documentation ! ! ! Further batch delay Reject (false negative?) HANDLING OF OOS RESULTS
- 30. Failure to investigate deviations 27% Incomplete investigations 25% Inadequate documentation and reporting 14% Inadequate corrective actions 11% Inadequate management review/oversight 9% Unjustified conclusions 9% Failure to assure timely investigation/closure 5% Failure investigation trends HANDLING OF OOS RESULTS
- 31. Prevention of OOS results - EQUIPMENT Prevention of OOS results - PEOPLE Training of Analysts, Supervisors, Lab Managers … We are all human… Periodic review of OOS/OOT/OOL results Regular review of investigations and of assignable causes of laboratory error with analysts Hands-on participation of analysts in investigations HANDLING OF OOS RESULTS
- 32. “ Operator Error” as stated reason for OOS results SHOULD BE SPECIFIC Operator Action – Inattention to Detail – Verbal or Written Communication Problem – Operator Monitoring Multiple Processes Operator Training: – Not Trained on Procedure – Not Trained on Current Version of Procedure – Insufficient Practice or Experience – Inadequate Content in Training Automation of routine laboratory tasks Reduce errors in dilution (serial dilutions) Reduce errors in weighing (especially multiple components) Improve reproducibility Improve safety Reduce stress level for analyst Reduce waiting time of sample (UV, O 2 , H 2 O, T o C …  degradation) HANDLING OF OOS RESULTS
- 33. NUMBER OF RETESTS HANDLING OF OOS RESULTS One analyst Two analysts, of at least the same level of experience ? Original test Investigation / Retesting One analyst, of higher level of experience ? 1 replicate Two tests in duplicates ? 2 replicates Two tests in triplicates ? 3 replicates Two tests in 4 replicates each ?
- 34. References and useful links 1. FDA: Guidance for Industry Investigating Out-of-Specification (OOS) Test Results for Pharmaceutical Production, Guidance for Industry, October 2006 ( http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm070287.pdf ) 2. PIC/S AIDE MEMOIRE ON INSPECTION OF QUALITY CONTROL LABORATORIES ( http://www.picscheme.org/publication.php?id=14 ) 3. MHRA presentations on OOS, July 2010 ( http://www.mhra.gov.uk/home/groups/comms-con/documents/websiteresources/con088214.pdf ) ( http://www.mhra.gov.uk/home/groups/comms-con/documents/websiteresources/con088215.pdf ) 4. United States of Americs, Plaintiff V. Barr Laboratories Inc., Defendants, Civil Suite 92-1744 ( http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/Manufacturing/UCM216425.pdf ) ( http://www.navigategmp.com/pdf/BarrLabs.pdf ) 5. Mettler-Toledo on-demand webinar by Dr. Joanne Ratcliff, Marketing Project Manager ( http://uk.mt.com/gb/en/home/events/webinar/ondemand/q_OoS_QAQC.html ) 6. Steven Kuwahara “A History of the OOS Problem”, BioPharm International, Vol.20, Issue 11 ( http://biopharminternational.findpharma.com/biopharm/article/articleDetail.jsp?id=470169 ) HANDLING OF OOS RESULTS
- 35. QUOTES FROM INSPECTORS … "If you want to keep us entertained, come up with inconclusive results“ “ Companies are very inventive in finding explanations to invalidate results” HANDLING OF OOS RESULTS
- 36. Thank you! Questions? Comments? HANDLING OF OOS RESULTS