




Phenylketonuria
- 1. Phenylketonuria a metabolic flowchart Kristine Joy Y. Sumanga MD-II CPU-ColMed
- 2. What is Phenylketonuria (PKU)? an autosomal recessive metabolic genetic disorder characterized by homozygous or compound heterozygous mutations in the gene for the hepatic enzyme phenylalanine hydroxylase (PAH), rendering it nonfunctional.[ This enzyme is necessary to metabolize the amino acid phenylalanine (Phe) to the amino acid tyrosine (Tyr).
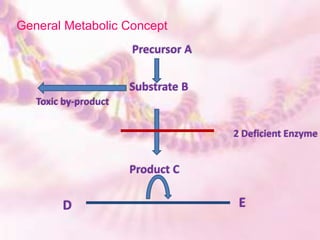
- 3. Precursor A Substrate B Toxic by-product Product C 2 Deficient Enzyme D E General Metabolic Concept
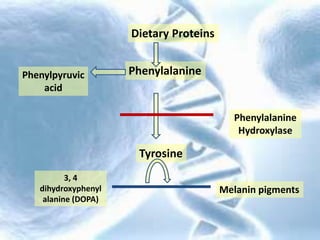
- 4. Dietary Proteins PhenylalaninePhenylpyruvic acid Tyrosine Phenylalanine Hydroxylase 3, 4 dihydroxyphenyl alanine (DOPA) Melanin pigments
- 5. The whole metabolic pathway of PKU
- 6. Phenylalanine Tyrosine p-hydroxyphenylpyruvic acid Homogentisic acid 2,5 dihydroxyphenylacetic acid Maleylacetoacetic acid Triiothyronine (T3) Thyroxine (T4) Glutamine GABA Pyruvate Oxaloacetate Glutamate decarboxylase Pyruvate carboxylase Phenylpyruvic acid Phenylalanine hydrolase Tyrosine transaminase p-hydroxyphenylpyruvic acid oxidase Homogentisic acid oxidase Fumanylacetoacetic acid Fumaric acid Acetoacetic acid Carbon dioxide + Water Iodinase 3,4 dihydroxyphenylalanine (DOPA) Melanin Pigments (decrease pigmentation, Blue eyes) (Cretinisim) (increased mousy odor) (seizure ,increase deep tendon reflex) (decrease ATP, mental retardation) (-)BH4 X X X X
- 7. Thank You and God bless