Prader Willi Syndrome ppt1
•Download as PPTX, PDF•
3 likes•2,364 views

Prader-Willi syndrome (PWS) is a genetic disorder that affects approximately 1 in 10,000-15,000 births. It results from a deletion or defect on chromosome 15 that causes specific genes to be inactive. Common features include low muscle tone, an insatiable appetite leading to obesity, developmental delays, and behavioral problems. Research has focused on understanding the causes of hypotonia and hyperphagia. While growth hormone treatment has led to benefits, the key genes responsible for features of PWS have not been identified, preventing targeted treatments. Identifying the causes of PWS could provide insights into other conditions like obesity and hypothalamic disorders.














Prader Willi Syndrome ppt1
- 1. I’m HUNGRY Roselaure Anstral Prader Willi Syndrome
- 2. What is Prader-Willi Syndrome Genetic Disorder  Prader-Willi-Labhart Syndrome (PWS) is the most common syndromic form of obesity and affects between 350,000 and 400,000 individuals worldwide. Both sexes are affected equally. Although prevalence estimates differ among studies, this is likely due to using different methods for case identification, and there is no strong evidence for increased risk in specific countries or gene pools. Within the United States, the rate of prevalence has been reported between 1 in 16,062 to 1 in 25,000 . Outside of the US, reported prevalence rates for PWS range from 1 per 8000 in rural Sweden to 1 per 16,000 in Western Japan, and a birth incidence of 1 per 27,000 in Flanders. Within the UK, a lower population prevalence of 1 in 52,000 was estimated, with a proposed true prevalence of 1 in 45,000.  Prevalence Rate: approx. 1 in 10,000 or 0.01% or 27,200 people in USA  “Failure of express of paternally inherited genes in the PWS region of chromosme 15” ~ Cassidy et al. 2009  Butler, MG, Hanchett, JM, Thompson, T. Clinical Findings and Natural History of Prader-willi Syndrome. In: Management of Prader-willi Syndrome, Butler, MG, Lee, PDK, Whitman, BY (Eds), Springer, New York 2006.  Burd L, Vesely B, Martsolf J, Kerbeshian J. Prevalence study of Prader-Willi syndrome in North Dakota. Am J Med Genet 1990; 37:97.  Butler MG. Prader-Willi syndrome: current understanding of cause and diagnosis. Am J Med Genet 1990; 35:319.  Akefeldt A, Gillberg C, Larsson C. Prader-Willi syndrome in a Swedish rural county: epidemiological aspects. Dev Med Child Neurol 1991; 33:715.  Ehara H, Ohno K, Takeshita K. Frequency of the Prader-Willi syndrome in the San-in district, Japan. Brain Dev 1995; 17:324.  Vogels A, Van Den Ende J, Keymolen K, et al. Minimum prevalence, birth incidence and cause of death for Prader-Willi syndrome in Flanders. Eur J Hum Genet 2004; 12:238.  Whittington JE, Holland AJ, Webb T, et al. Population prevalence and estimated birth incidence and mortality rate for people with Prader-Willi syndrome in one UK Health Region. J Med Genet 2001; 38:792.
- 3. 3 Prader-Willi Syndrome • In 1956, PWS was identified as a constellation of symptoms by Swiss physicians A. Prader, H. Willi and A. Labhart • PWS is a lifelong, life-threatening, non-inherited genetic disorder that results from a defect on Chromosome 15 • PWS occurs in 1 in 12,000-15,000 births, or approximately 25,000 people in U.S. Of these, 75-80% are either undiagnosed or unknown to PWSA(USA). • In NV, there are only 39 known cases of PWS; 13 of which are in the greater Las Vegas area. There are an additional 70-75% either undiagnosed or unknown to PWSA(USA). • PWS equally affects all races and both sexes.
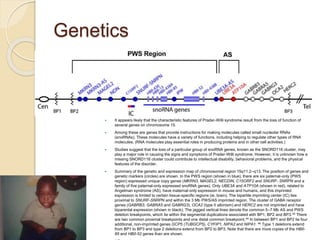
- 4. Genetics  It appears likely that the characteristic features of Prader-Willi syndrome result from the loss of function of several genes on chromosome 15.  Among these are genes that provide instructions for making molecules called small nucleolar RNAs (snoRNAs). These molecules have a variety of functions, including helping to regulate other types of RNA molecules. (RNA molecules play essential roles in producing proteins and in other cell activities.)  Studies suggest that the loss of a particular group of snoRNA genes, known as the SNORD116 cluster, may play a major role in causing the signs and symptoms of Prader-Willi syndrome. However, it is unknown how a missing SNORD116 cluster could contribute to intellectual disability, behavioral problems, and the physical features of the disorder.  Summary of the genetic and expression map of chromosomal region 15q11.2–q13. The position of genes and genetic markers (circles) are shown. In the PWS region (shown in blue), there are six paternal-only (PWS region) expressed unique copy genes (MKRN3, MAGEL2, NECDIN, C15ORF2 and SNURF- SNRPN and a family of five paternal-only expressed snoRNA genes). Only UBE3A and ATP10A (shown in red), related to Angelman syndrome (AS), have maternal-only expression in mouse and humans, and this imprinted expression is limited to certain tissue-specific regions (ie, brain). The bipartite imprinting center (IC) lies proximal to SNURF-SNRPN and within the 3 Mb PWS/AS imprinted region. The cluster of GABA receptor genes (GABRB3, GABRA5 and GABRG3), OCA2 (type II albinism) and HERC2 are not imprinted and have biparental expression (shown in black). The jagged vertical lines denote the common 5–7 Mb AS and PWS deletion breakpoints, which lie within the segmental duplications associated with BP1, BP2 and BP3.45 There are two common proximal breakpoints and one distal common breakpoint.44 In between BP1 and BP2 lie four additional, non-imprinted genes GCP5 (TUBGCP5), CYFIP1, NIPA2 and NIPA1. 46 Type 1 deletions extend from BP1 to BP3 and type 2 deletions extend from BP2 to BP3. Note that there are more copies of the HBII- 85 and HBII-52 genes than are shown.
- 5. Frequencies ÔÇó Partenal ÔÇó Maternal Unipatental disomy ÔÇó Imprinting Defect Causes of Prader-Willi syndrome (PWS). This diagram represents chromosomes 15 of an unaffected father (blue with PWS critical region highlighted) and unaffected mother (pink). The children represent the three different mechanisms for PWS: a deletion in the PWS region on chromosome 15 that is inherited from the father (present in about 70 percent of patients) (A); both chromosomes 15 are inherited from the mother and the PWS region from the father is missing (present in about 25 percent of patients) (B); and a defect in methylation inherited from the father (present in less than 5 percent of patients) (C). In this case, the genes in the PWS critical region on the chromosome 15 inherited from the father are inactivated, similar to those of the mother.
- 7. Three Phases of PWS ÔÇó1.) Hypotonic Stage (prenatal-infancy) ÔÇó2.) Hyperphagic Stage (childhood) ÔÇó3.) Adolescence and Adulthood
- 8. Research History ÔÇó Origianl Research Focus: weak muscle tone (hypotonia) Infantile hypotonia is a nearly universal finding, causing decreased movement and lethargy with decreased spontaneous arousal, weak cry, and poor reflexes, including a poor suck. The hypotonia is central in origin, and neuromuscular studies including muscle biopsy, when done for diagnostic purposes, are generally normal or show nonspecific signs of disuse. The poor suck and lethargy result in failure to thrive in early infancy, and gavage feeding or the use of special nipples is generally required for a variable period of time, usually weeks to months.4 By the time that the child is drinking from a cup or eating solids, a period of approximately normal eating behavior occurs. The hypotonia improves over time, but adults remain mildly hypotonic with decreased muscle bulk and tone.
- 9. Normal Muscle Fiber Nucleus (N) Z line (Z), A and I bands (A, I), mitochondria (M) and a lipid droplet (L). Arror: sarcroplasmic reticulum profile Afifi AK, Zellweger H. Pathology of muscular hypotonia in the Pradwer-Willi syndrome. J Neuro Sci 1969; 4: 46- 61
- 10. Z line tortuosity (twisted/ crookedness) and irregularity, myofilementous disarray and disruption of sarcomeral structure, intact myofibril, intact Z line Afifi AK, Zellweger H. Pathology of muscular hypotonia in the Pradwer-Willi syndrome. J Neuro Sci 1969; 4: 46-61
- 12. Living With PWS
- 13. ÔÇó Growth hormone replacement in PWS has resulted in dramatic benefits to the phenotype, health and self-image of those treated. But much is yet to be learned about this complex disorder. Clinically, the most urgent need is arguably to identify the cause of hyperphagia in hopes of finding pharmacologic agents that can diminish its impact. Would the effect of such a drug be to improve behavior and cognitive ability as well as health? If obesity could be more easily avoided, the major cause of morbidity and mortality would be eliminated and significantly improved health and quality of life could be expected. ÔÇó It has been 27 years since the genetic region responsible for PWS was first identified; yet we still do not know the precise gene(s) responsible for the phenotype. Recent data suggests a key role for the HBII-85 snoRNA gene,48, 73 and the near future should uncover its gene targets and hopefully lead to treatments. Identification of the responsible gene(s) may also be extremely instructive concerning causes of obesity, hypogonadism, other hypothalamic deficiencies, psychosis, and autism in the general population. PWS has already taught us much about imprinting, and may have much more to teach us
- 14. Thank You
- 15. References: • Afifi AK, Zellweger H. Pathology of muscular hypotonia in the Pradwer-Willi syndrome. J Neuro Sci 1969; 4: 46- 61 • Butler, M.G. and Thompson, T. (2000) Prader-Willi Syndrome: Clinical and Genetic Findings. The Endocrinologist 10 (4) Suppl 1:3S-16S • Cassidy, S.B. and Schwartz, S. (1998) Prader-Willi and Angelman Syndromes: Disorders of Genomic Imprinting. Medicine 77: 140-151. • Cassidy, SB and Driscoll, DJ. (2009). Prader–Willi syndrome. European Journal of Human Genetics 17, 3– 13; doi:10.1038/ejhg.2008.165; published online 10 September 2008 • http://www.nature.com/ng/journal/v40/n6/abs/ng.158. html