


















![Correlation coefficient
True positive
p╬▒
False positive
(overpredicted)
o╬▒
True negative
n╬▒
False negative
(underpredicted)
u╬▒
])
][
][
[
]
([ ’üĪ
’üĪ
’üĪ
’üĪ
’üĪ
’üĪ
’üĪ
’üĪ
’üĪ
’üĪ
’üĪ
’üĪ
’üĪ o
p
u
p
o
n
u
n
o
u
n
p
C ’Ć½
’Ć½
’Ć½
’Ć½
’ĆĮ
’āŚ
’ĆŁ
’āŚ
Ca = 1 (=100%)](https://image.slidesharecdn.com/proteinstructureprediction-240914174752-57cbf7d0/85/protein-structure-prediction-in-bioinformatics-ppt-21-320.jpg)









































More Related Content
Similar to protein structure prediction in bioinformatics.ppt (20)
More from DrSudha2 (11)









Recently uploaded (20)




















protein structure prediction in bioinformatics.ppt
- 2. Prediction in bioinformatics Important prediction problems: ’āśProtein sequence from genomic DNA. ’āśProtein 3D structure from sequence. ’āśProtein function from structure. ’āśProtein function from sequence.
- 3. From DNA to Cell Function DNA sequence (split into genes) AminoAcid Sequence Protein 3D Structure Protein Function Cell Activity codes for folds into dictates determines has MNIFEMLRID EGLRLKIYKD TEGYYTIGIG HLLTKSPSLN AAKSELDKAI GRNCNGVITK DEAEKLFNQD VDAAVRGILR NAKLKPVYDS LDAVRRCALI NMVFQMGETG VAGFTNSLRM LQQKRWDEAA VNLAKSRWYN QTPNRAKRVI TTFRTGTWDA YKNL ?
- 4. Protein structure: Limitations ŌĆó Not all proteins or parts of proteins assume a well-defined 3D structure in solution. ŌĆó Protein structure is not static, there are various degrees of thermal motion for different parts of the structure. ŌĆó There may be a number of slightly different conformations in solution. ŌĆó Some proteins undergo conformational changes when interacting with certain substances. ŌĆó Expected best residue-by-residue accuracies for secondary structure prediction from multiple protein sequence alignment. ŌĆó To address detailed functional biological questions.
- 5. Experimental Protein Structure Determination ŌĆó X-ray crystallography ŌĆō the most advanced method available for obtaining high-resolution structural information about biological macromolecules ŌĆō in vitro ŌĆō needs crystals ŌĆō ~$100-200K per structure ŌĆó NMR ŌĆō fairly accurate ŌĆō in vivo ŌĆō no need for crystals ŌĆō limited to very small proteins ŌĆó Cryo-electron-microscopy ŌĆō imaging technology ŌĆō low resolution
- 6. Why predict protein structure? ŌĆó Over millions known sequences, 1,25,309 known structures. ŌĆó Structural knowledge brings understanding of function and mechanism of action. ŌĆó Predicted structures can be used in structure-based drug design. ŌĆó It can help us understand the effects of mutations on structure and function. ŌĆó To analyze sequence structure gap. ŌĆó Can help in prediction of function. ŌĆó It is a very interesting scientific problem-50 years effort. ŌĆó Prediction in one dimension ŌĆō Secondary structure prediction ŌĆō Surface accessibility prediction
- 7. ŌĆó Historically first structure prediction methods predicted secondary structure. ŌĆó Can be used to improve alignment accuracy. ŌĆó Can be used to detect domain boundaries within proteins with remote sequence homology. ŌĆó Often the first step towards 3D structure prediction. ŌĆó Informative for mutagenesis studies. Secondary structure prediction
- 8. Predicting Secondary Structure From Primary Structure ŌĆó accuracy 64-75%. ŌĆó higher accuracy for a-helices than for b-sheets. ŌĆó accuracy is dependent on protein family. ŌĆó predictions of engineered (artificial) proteins are less accurate. Assumptions ŌĆó The entire information for forming secondary structure is contained in the primary sequence. ŌĆó Side groups of residues will determine structure. ŌĆó Examining windows of 13-17 residues is sufficient to predict secondary structure . -╬▒-helices 5ŌĆō40 residues long -╬▓-strands 5ŌĆō10 residues long
- 9. Why Secondary Structure Prediction? ŌĆó Simply easier problem than 3D structure prediction. ŌĆó Accurate secondary structure prediction can be an important information for the tertiary structure prediction. ŌĆó Improving alignment accuracy. ŌĆó Protein function prediction. ŌĆó Protein classification.
- 10. Protein structure prediction ŌĆó The inference of the three-dimensional structure of a protein from its amino acid sequence. ŌĆō i.e. the prediction of its folding and its secondary and tertiary structure from its primary structure. ŌĆó Structure prediction is fundamentally different from the inverse problem of protein design. ŌĆó Protein structure prediction is one of the most important goals pursued by bioinformatics and theoretical chemistry. ŌĆó It is highly important in medicine (in drug design) and biotechnology (in the design of novel enzymes).
- 11. Methods of structure prediction ’āśAb initio protein folding approaches ’āśComparative (homology) modelling ’āśFold recognition/threading
- 12. History of protein secondary structure prediction ’āśFirst generation ’āśBased on single residue statistics. ’āśExample: Chou-Fasman method, LIM method, GOR I, etc ’āśAccuracy: low ’āśSecondary generation ’āśBased on segment statistics. ’āśExamples: ALB method, GOR III, etc ’āśAccuracy: ~60% ’āśThird generation ’āśBased on long-range interaction, homology based ’āśExamples: PHD ’āśAccuracy: ~70%
- 13. First generation methods: single residue statistics Chou & Fasman (1974 & 1978) : ’é¦ Some residues have particular secondary-structure preferences. ’é¦ Based on experimental frequencies of residues in ’üĪ-helices, ’üó-sheets, and coils. Examples: Glu ╬▒-helix Val ╬▓-strand ’é¦ Accuracy ~50 - 60% Q3
- 14. Chou-Fasman statistics ŌĆó R ŌĆō amino acid, S- secondary structure ŌĆó f(R,S) ŌĆō number of occurrences of R in S ŌĆó Ns ŌĆō total number of amino acids in conformation S ŌĆó N ŌĆō total number of amino acids ŌĆó P(R,S) ŌĆō propensity of amino acid R to be in structure S ŌĆó P(R,S) = (f(R,S)/f(R))/(Ns/N)
- 15. Example ŌĆó #residues=20,000, ŌĆó #helix=4,000, ŌĆó #Ala=2,000, ŌĆó #Ala in helix=500 ŌĆó f(Ala, ) = 500/20,000, ╬▒ ŌĆó f(Ala) = 2,000/20,000 ŌĆó p( ) = / =4,000/20,000 ╬▒ ╬Ø╬▒ ╬Ø ŌĆó P = (500/2000) / (4,000/20000) = 1.25
- 16. Second generation methods: segment statistics ŌĆó Similar to single-residue methods, but incorporating additional information (adjacent residues, segmental statistics). ŌĆó Problems: ŌĆō Low accuracy - Q3 below 66% (results). ŌĆō Q3 of ’üó-strands (E) : 28% - 48%. ŌĆō Predicted structures were too short.
- 17. The GOR method ŌĆó Developed by Garnier, Osguthorpe & Robson ŌĆó Build on Chou-Fasman Pij values ŌĆó Evaluate each residue PLUS adjacent 8 N-terminal and 8 carboxyl-terminal residues ŌĆó Sliding window of 17 residues. ŌĆó underpredicts b-strand regions ŌĆó GOR method accuracy Q3 = ~64%
- 18. Third generation methods ŌĆó Third generation methods reached 77% accuracy. ŌĆó They consist of two new ideas: 1. A biological idea ŌĆō Using evolutionary information based on conservation analysis of multiple sequence alignments. 2. A technological idea ŌĆō Using neural networks.
- 19. Artificial Neural Networks An attempt to imitate the human brain (assuming that this is the way it works).
- 20. Neural network models - machine learning approach - provide training sets of structures (e.g. a-helices, non a -helices) - computers are trained to recognize patterns in known secondary structures - provide test set (proteins with known structures) - accuracy ~ 70 ŌĆō75%
- 21. Correlation coefficient True positive p╬▒ False positive (overpredicted) o╬▒ True negative n╬▒ False negative (underpredicted) u╬▒ ]) ][ ][ [ ] ([ ’üĪ ’üĪ ’üĪ ’üĪ ’üĪ ’üĪ ’üĪ ’üĪ ’üĪ ’üĪ ’üĪ ’üĪ ’üĪ o p u p o n u n o u n p C ’Ć½ ’Ć½ ’Ć½ ’Ć½ ’ĆĮ ’āŚ ’ĆŁ ’āŚ Ca = 1 (=100%)
- 22. Reasons for improved accuracy ŌĆó Align sequence with other related proteins of the same protein family. ŌĆó Find members that has a known structure. ŌĆó If significant matches between structure and sequence assign secondary structures to corresponding residues.
- 23. New and Improved Third-Generation Methods Exploit evolutionary information. Based on conservation analysis of multiple sequence alignments. ŌĆó PHD (Q3 ~ 70%) Rost B, Sander, C. (1993) J. Mol. Biol. 232, 584-599. ŌĆó PSIPRED (Q3 ~ 77%) Jones, D. T. (1999) J. Mol. Biol. 292, 195-202. Arguably remains the top secondary structure prediction method.
- 24. Secondary Structure Prediction Summary 1st Generation - 1970s ŌĆó Q3 = 50-55% ŌĆó Chou & Fausman, GOR 2nd Generation -1980s ŌĆó Q3 = 60-65% ŌĆó Qian & Sejnowski, GORIII 3rd Generation - 1990s ŌĆó Q3 = 70-80% ŌĆó PhD, PSIPRED Many 3rd+ generation methods exist: PSI-PRED - http://bioinf.cs.ucl.ac.uk/psipred/ JPRED - http://www.compbio.dundee.ac.uk/~www-jpred/ PHD - http://www.embl-heidelberg.de/predictprotein/predictprotein.html NNPRED - http://www.cmpharm.ucsf.edu/~nomi/nnpredict.html
- 25. Protein 3D structure data ’āśThe structure of a protein consists of the 3D (X,Y,Z) coordinates of each non-hydrogen atom of the protein. ’āśSome protein structure also include coordinates of covalently linked prosthetic groups, non-covalently linked ligand molecules, or metal ions. ’āśFor some purposes (e.g. structural alignment) only the C╬▒ coordinates are needed. Example of PDB format: X Y Z occupancy / temp. ATOM 18 N GLY 27 40.315 161.004 11.211 1.00 10.11 ATOM 19 CA GLY 27 39.049 160.737 10.462 1.00 14.18 ATOM 20 C GLY 27 38.729 159.239 10.784 1.00 20.75 ATOM 21 O GLY 27 39.507 158.484 11.404 1.00 21.88 Note: the PDB format provides no information about connectivity between atoms. The last two numbers (occupancy, temperature factor) relate to disorders of atomic positions in crystals.
- 27. ’āśBuilding a protein structure model from X-ray data ’āśBuilding a protein structure model from NMR data ’āśComputing the energy for a given protein structure (conformation) ’āśEnergy minimization: Finding the structure with the minimal energy according to some empirical ŌĆ£force fieldsŌĆØ. ’āśSimulating the protein folding process (molecular dynamics) ’āśStructure visualization Structure visualization ’āśComputing secondary structure from atomic coordinates ’āśProtein superposition, structural alignment Protein superposition, structural alignment ’āśProtein fold classification Protein fold classification ’āśThreading: finding a fold (prototype structure) that fits to a sequence Threading: finding a fold (prototype structure) that fits to a sequence ’āśDocking: fitting ligands onto a protein surface by molecular dynamics or energy minimization ’āśProtein 3D structure prediction from sequence Protein 3D structure prediction from sequence Protein structure: Some computational tasks Protein structure: Some computational tasks
- 28. Viewing protein structures When looking at a protein structure, we may ask the following types of questions: ŌĆó Is a particular residue on the inside or outside of a protein? ŌĆó Which amino acids interact with each other? ŌĆó Which amino acids are in contact with a ligand (DNA, peptide hormone, small molecule, etc.)? ŌĆó Is an observed mutation likely to disturb the protein structure? Standard capabilities of protein structure software: ŌĆó Display of protein structures in different ways (wireframe, backbone, sticks, spacefill, ribbon. ŌĆó Highlighting of individual atoms, residues or groups of residues ŌĆó Calculation of interatomic distances ŌĆó Advanced feature: Superposition of related structures
- 29. Example: c-abl oncoprotein SH2 domain, display wireframe
- 30. Example: c-abl oncoprotein SH2 domain, display sticks
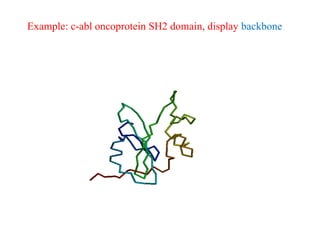
- 31. Example: c-abl oncoprotein SH2 domain, display backbone
- 32. Example: c-abl oncoprotein SH2 domain, display spacefill
- 33. Example: c-abl oncoprotein SH2 domain, display ribbons
Editor's Notes
- #20: Simulate the brain. Selection of training sets is extremely important. Different protein families, only one or two representative from each family.