Tifa site directed mutagenesis
ŌĆóDownload as PPTX, PDFŌĆó
2 likesŌĆó1,356 views

Mutagenesis terarah pada DNA.. rekayasa genetika untuk melakukan perubahan secara sengaja dan spesifik pada daerah nukleotida tertentu dari sekuens DNA yang diinginkan
























Tifa site directed mutagenesis
- 1. Site-directed Mutagenesis By Tifa Rachmi Kusumastuti 21112026
- 2. Mutagenesis? ’éŚ Proses dimana mutagen menyebabkan sekuens DNA / informasi genetik organisme mengalami perubahan yang menghasilkan mutasi. ’éŚ Dapat terjadi secara spontan di alam atau buatan melalui proses laboratorium.
- 4. Site-directed Mutagenesis ’éŚ ŌĆ£Instead of crudely mutagenizing many cells or organisms and then analyzing many thousands or million of off-springs to isolate a desired mutant, it is now possible to change specifically any given base in a cloned sequence. This technique is known as Site ŌĆō Directed MutagenesisŌĆØ (Primrose)
- 5. Site-directed Mutagenesis ’éŚ Metode untuk melakukan perubahan secara sengaja dan spesifik pada daerah nukleotida tertentu dari sekuens DNA yang diinginkan biasanya untuk mempelajari fungsi protein. ’éŚ Perubahan pada basa dapat berupa single mutation (point mutation) atau multiple base change (delesi atau insersi). ’éŚ Site Direction Mutagenesis : ’ü▒ KunkelŌĆÖs Method ’ü▒ Cassette Mutagenesis ’ü▒ Primer Extention (single-primer method) ’ü▒ PCR for Site-directed Mutagenesis
- 6. Sejarah Penemuan ’éŚ 1920 Herman Muller menemukan mutasi pada lalat buah akibat pengaruh paparan sinar X-rays ’éŚ 1971 Clyde Hutchison and Marshall Edgell membuat mutasi menggunakan fragmen kecil dari phage ŽĢX174 dan restriction nucleases. ’éŚ 1973 Charles Weissmann menggunakan N4- hydroxycytidine yang mengubah GC menjadi AT. ’éŚ 1978 Clyde Hutchison and Michael Smith mendeskripsikan site-directed mutagenesis pertama kalinya menggunakan oligonukleotida sebagai dalam metode primer ekstensi. ’éŚ 1993 Michael Smith menerima hadiah nobel dalam bidan kimia bersam Kary B. Mullis (yang mengembangkan metode PCR).
- 7. Mekanisme Umum Site-directed Mutagenesis ’éŚ Primer DNA (sintetik) mutasi sesuai keinginan dan merupakan komplementer dari DNA template daerah yang akan dimutasi. ’éŚ DNA polymerase akan melakukan ekstensi primer. ’éŚ DNA yang mengandung daerah mutasi ini ditransformasikan pada sel inang sebagai vektor untuk selanjutnya dilakukan proses kloning dan seleksi.
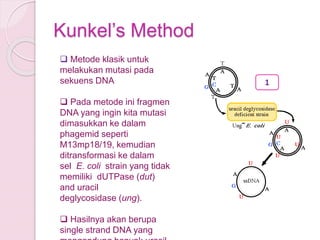
- 8. KunkelŌĆÖs Method ’ü▒ Metode klasik untuk melakukan mutasi pada sekuens DNA ’ü▒ Pada metode ini fragmen DNA yang ingin kita mutasi dimasukkan ke dalam phagemid seperti M13mp18/19, kemudian ditransformasi ke dalam sel E. coli strain yang tidak memiliki dUTPase (dut) and uracil deglycosidase (ung). ’ü▒ Hasilnya akan berupa single strand DNA yang 1
- 9. ’ü▒ Single strand DNA ini kemudian diekstrak dan dijadikan sebagai cetakan (template) untuk mutagenesis. ’ü▒ DNA diinkubasi bersama mutagenic oligonukleotida yang mengandung titik mutasi yang diinginkan sebagai primer extensi. ’ü▒ DNA akan membentuk double strand dalam mixture Klenow, dNTPs, Ligase dan ATP. Hasilnya berupa double strand DNA yang pada strand baru mengandung ŌĆśŌĆØTŌĆØ dan strand lama mengandung ŌĆ£UŌĆØ. 2
- 10. ’ü▒ DNA hibrid ditransformasikan kedalam E. coli strain yang membawa gen wild type dut and ung. ’ü▒ DNA lama yang mengandung ŌĆ£UŌĆØ akan didegradasi dan diganti dengan copy baru yang mengandung ŌĆ£TŌĆØ. ’ü▒ Pada akhirnya yang tersisa hanya DNA hasil mutasi. 3
- 12. Cassette Mutagenesis ’ü▒ Metode Cassette tidak membutuhkan proses pemanjangan primer oleh DNA polymerase seperti metode lain. ’ü▒ Fragmen DNA disintesis dan dimasukkan ke dalam plasmid. ’ü▒ Melibatkan Enzim restriksi untuk memotong pada derah target sekuens yang diinginkan. ’ü▒ Fragmen DNA yang mengandung oligonukleotida gen interest disambung pada daerah restriksi menggunakan enzim ligase. ’ü▒ Keuntungan : efisiensi dalam menghasilkan mutan hampir 100%, lebih murah karena tidak membutuhkan primer, Note : limited by the availability of suitable restriction sites flanking the site that is to be mutated.
- 13. Kodon target mutasi dipotong menggunakan enzim restriksi
- 14. Primer Extention Method 1. Single strand DNA rekombinan berasal dari bacteriophage M13 2. Primer mutagenic oligonukleotida (didesain dan dibuat memiliki mutasi yang diinginkan) 3. Hibridisasi primer dengan mutagenic oligonucleotida dengan target DNA 4. Ekstensi primer oleh DNA polymerase 5. Transfeksi atau ditransformasikan ke bakteri 6. Screening 7. Sekuensing
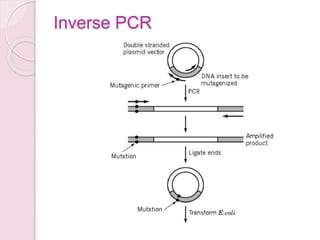
- 22. Inverse PCR
- 24. Whole Plasmid Mutagenesis ’éŚ "Site-directed mutagenesis in one day with >80% efficiency.". Strategies 9 (3): 3ŌĆō4. ’éŚ pfu polymerase ’éŚ Restriction enzyme such as DpnI
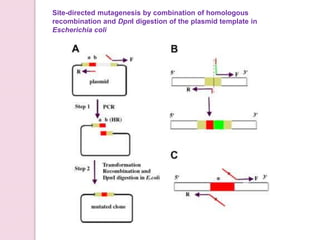
- 27. Site-directed mutagenesis by combination of homologous recombination and DpnI digestion of the plasmid template in Escherichia coli
- 28. Manfaat Site-directed Mutagenesis ’ü▒ Sebagai alat untuk mempelajari fungsi dan properti dari sekuens DNA tertentu karena mutasi yang dibuat secara spesifik. ’ü▒ Memungkinkan kita dapat mengetahui dengan detail identitas dan fungsi dari suatu asam amino dan melihat efek aktivitas enzimatiknya. ’ü▒ Membuat protein yang dirancang untuk kebutuhan tertentu.
Editor's Notes
- dUTPase┬Ā(dut) and┬Āuracil deglycosidase┬Ā(ung). Both enzymes are part of a DNA repair pathway that protects the bacterial chromosome from mutations by the spontaneous deamination of dCTP to dUTP. The dUTPase deficiency prevents the breakdown of dUTP, resulting in a high level of dUTP in the cell. The uracil deglycosidase deficiency prevents the removal of uracil from newly-synthesized DNA. As the double-mutant┬ĀE. coli┬Āreplicate the phage DNA, its enzymatic machinery may therefore misincorporates dUTP instead of dTTP, resulting in single stranded DNA which contains some uracils (ssUDNA).
- Usually the restriction enzymes that cuts at the plasmid and the oligonucleotide are the same, permitting sticky ends of the plasmid and insert to ligate to one another.
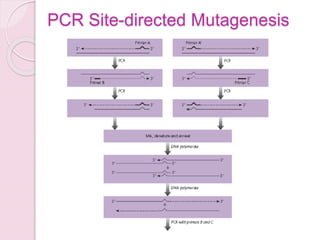
- Site-Directed Mutagenesis by Traditional PCR.┬ĀPrimers incorporating the desired base changes are used in PCR. As the primers are extended, the mutation is created in the resulting amplicon.
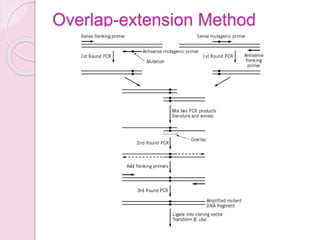
- The overlap-extension method requires four┬Āoligonucleotide primers and three separate amplification reactions (34, 35). Two complementary mutagenic primers induce the mutation into the desired sequence of DNA, and two flanking primers amplify the mutant fragment and facilitate cloning of the PCR fragment into a suitable vector. By this approach (Fig. 3), a variety of mutations can be created, such as single base-pair changes, deletions, and insertions. First, two separate PCR reactions are set up in parallel. One reaction has the "sense" mutant primer and an "anti-sense" flanking primer 3ŌĆ▓ to the mutation site. The other reaction contains the anti-sense mutant primer and the sense flanking primer 5ŌĆ▓ to the mutation site. The two amplified fragments contain mutations at the 5ŌĆ▓ or 3ŌĆ▓ terminus, respectively. In the second round of amplification, the two fragments from the first round of PCR are purified, then used as templates for amplification using only the flanking primers. After amplification, the mutation is contained within the target DNA segment, which is cloned into appropriate vectors for DNA sequencing and subsequent functional studies. Figure 3. Site-directed mutagenesis by overlap-extension PCR. The two first rounds of PCR produced two overlapping fragments of the original template, both containing the mutation within the overlap region. These two PCR products are annealed and then subjected to a second round of PCR to generate the entire fragment with the mutation. The flanking primers contain the restriction sites for ligating the fragment back into the original vector.
- The megaprimer method uses only a single mutagenic┬Āprimer to create mutations in the target template (36-38) (Fig. 4). In the first round of amplification, the wild-type template is amplified using either a sense or anti-sense mutagenic primer and an appropriate flanking primer. The amplified product is then used in a second round of PCR with wild-type template and the other flanking primer to create a fragment of the same length as the original target DNA containing the desired mutation. The key to this method is that the amplified product from the first round of PCR is used as a primer in the second round of PCR. Compared to the four-primer method, this procedure requires only a single mutagenic primer and yields more of the full-length product. This is probably due to the instability of the 10- to 20-basepair overlap between the two mutant templates during the second round of amplification when using the four-primer method. In the megaprimer method, the overlap between the template and mutagenic strands is more extensive. Figure 4. Site-directed mutagenesis by the megaprimer PCR method. The first round of PCR is used to make a fragment of the template DNA containing the desired mutation. This "megaprimer" is then hybridized to the wild-type template DNA, and a second round of PCR is carried out, to generate the entire molecule with the mutation. The flanking primers contain the restriction sites for cloning the fragment back into the vector
- A third approach, termed inverse PCR, uses only two primers to create the desired mutation (39) (Fig. 5). The key feature of this method is that in making the mutation, the entire vector is amplified. The two primers, one containing the desired mutation, extend on the circular template DNA in opposite directions. Amplification ultimately yields a linear, double-stranded DNA molecule containing the mutation at one end. Following amplification, the ends are ligated, and the resulting circular DNA molecule is transformed into E. coli. There are a number of variations of this method that improve the efficiency of mutagenesis (reviewed in 33). Figure 5. Site-directed mutagenesis by inverse PCR. The vector is cleaved by a restriction enzyme at a single site, close to the site to be mutated. Two primers from the two ends of the linearized molecule, one containing the desired mutation, are used to amplify the linear molecule and produce molecules with the mutation. After ligating the ends to regenerate a circular molecule, the vector is used to transform E. coli.
- Figure 3. Site-Directed Mutagenesis by Inverse PCR. The primers used are 5ŌĆÖ-phosphorylated to allow ligation of the amplicon ends after PCR. A high fidelity DNA polymerase that creates blunt-ended products is used for the PCR to produce a linearized fragment with the desired mutation, which is then recircularized by intramolecular ligation. (A) Deletion: Primers that hybridize to regions on either side of the area to be deleted are used. (B) Substitution: One of the primers contains the desired mutation (blue bubble). (C) Insertion: The primers hybridize to regions on either side of the location of the desired insertion (black, dotted line). One primer contains the additional sequence that will be inserted (blue line).
- Fig. 1.┬ĀSchematic diagram of the site-directed mutagenesis method that combines homologous recombination and┬ĀDpnI digestion of the templates in┬ĀE. coli┬ĀBUNDpnI. (A) Protocol for sequence deletion. The target sequence for deletion is denoted in gray, while the flanking regions are denoted in light brown and red. The regions of primers that can hybridize on each strand of the template to gene-specific sequence immediate to the deletion are indicated by arrows, respectively. Fifteen nucleotides immediately 5ŌĆ▓ to the region to be deleted (indicated by ab) should be contained in primer F. Step 1: PCR amplification. The desired mutation(s)-containing DNA molecules with a homologous region at the terminal ends were created by PCR. Step 2: Transformation. The mutated plasmid was efficiently generated on direct transformation of the amplified PCR products into┬ĀE. coli┬ĀBUNDpnI. (B) Sequence insertion. As for sequence insertion, to simplify the figure, only the initial primer binding sites and the final inserted products are depicted. The insertion is denoted by a red and green rectangle. The gene-specific regions of primers that can hybridize on each strand of the template are located immediate to the site of the insertion, respectively. The sequence to be inserted is incorporated at the 5ŌĆ▓ end of primer F. Primer R contains a 5ŌĆ▓ tail complementary to the 15 outermost nucleotides of primer F (indicated by a red line). The final product with the inserted sequence denoted by the red and green rectangles was obtained after transformation. (C) Point mutations. Only the initial primer binding sites are shown. The region to be mutated is represented by a red rectangle, with the target site indicated by an asterisk. The desired nucleotides (indicated by black square dots) to be introduced into the target sequence are incorporated into the overlapping region of primers. The products carrying the designed mutations are generated by PCR amplification as described for sequence deletion.