20130609 Wako.R •»•‘•√•Ø•‚•«•Î§Ú”√§§§ø•Ð©`•´•Ì•§•…òS«˙§Œ¡˜––Ω‚Œˆ
13 likes3,010 views

–°’hº“§À§ §Ì§¶§ŒDTMΩ‚Œˆ§Œ•—•Ø•Í§¿§»Àº§√§∆§‚§Ω§Ï—‘§¶§ŒΩ˚÷𧫰£ http://seikichi.hatenablog.com/entry/2013/04/29/013608 http://moon.kmc.gr.jp/~seikichi/spring2013/kmc-spring-camp-2013.pdf




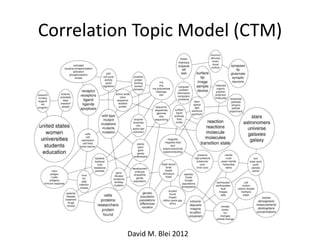
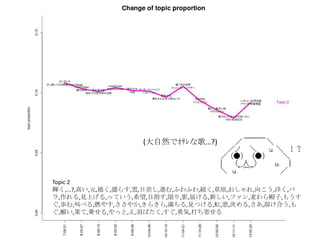
![Correlation Topic Model (CTM)
LDA§œ∏˜•»•‘•√•Ø§¨∂¿¡¢§¿§»Àº§√§∆§§§Î°£
°∏ÕÍ»´§À∂¿¡¢§∑§ø•»•‘•√•Ø°π§œ¨Fågµƒ§«§œ§ §§°£
•»•‘•√•Ø§Ú¡¢§∆§π§Æ§Î§»•¿•·§À§ §Î°£
•»•‘•√•ØÈg§ŒÈvÇS§Úøºë]
David M. Blei 2012](https://image.slidesharecdn.com/20130609wakor2-130728054542-phpapp02/85/20130609-Wako-R-13-320.jpg)





![Topic 6
•ø,“‚Œ∂,§»§´,§´§¥,∑÷§´§Î,—®,ö𧨧ƒ§Ø,se,§ §Û§´,control,¥Û«–,∂»,øº§®§Î,§Ω§Ï§«§‚,§ø§Í,§‰,À¿§Ã,πƒÑ”,§π§Æ§Î,§¡§„
§¶,öð∑÷,§Ø§È§§,•´• ,°Ó),§∏§„§¶,¬Ñ§Ø,œ”,∫ÙŒ¸,öð§≈§Ø,§≥§π,§´§‚,•Œ,ƒÐ¡¶,þ`§¶,팧∆§Î,§∑,È]§∏§Î,Œƒ◊÷,•þ•√•Ø•π,–—§·
§Î,)°π,§¿§±§…,÷ŸÈg,§Ð§Î,Õ¥§þ,§¡§„,•≠•È•§,»þ’Ñ,è䧧,”˚§∑§§
Topic 8
Ū§®§Î,!!!!,“ªæw,ƒ–,§»§≠,ª·§¶,”◊§§,»º§®§Î,ª·§®§Î,Õ®§Íþ^§Æ§Î,Ω~åù,Àÿî≥,÷€,§µ§Ë§¶§ §È,≥Øüܧ±,Ó䧶,±Ø§∑§§,≥ˆª·
§¶,—‘§®§Î,...?,Àº§§,ÓÆ,Ö€§Ø,∂»,§Ω§Ì§Ω§Ì,þ^§Æ§Î,∞¬,±À,“䧃§·§Î,æA§Ø,ºæπù,«–§ §§,öi§ý,∞K§π,ÈvÇS,§≥§Ï§´§È,Ωϧ±,§…
§Û§ ,¶§È§π,Ó䧧,é◊§ƒ,òS§∑§§,§™§∑§„§Ï,?,¬√¡¢§ƒ,“Á§Ï§Î, À≤ð,ºs ¯,»À–Œ,?
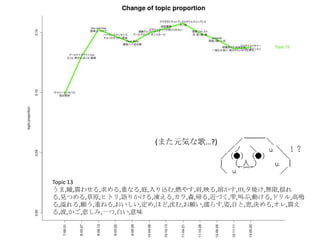
Topic 13
§¶§Þ,Õ´,’§Ô§ª§Î,«Û§·§Î,÷ÿ§ §Î,Õ•,»Î§ÍÞz§ý,»º§‰§π,ºÁ,”≥§Î,»Ð§´§π,!!!,œ¶üܧ±,üoœÞ,ìe§Ï§Î,“䧃§·§Î,≤ð‘≠,•“•»
•Í,’Z§Í§´§±§Î,Ɉ§®§Î,•´•È,…≠,颧Î,Ω¸§≈§Ø,Î~,Ω–§÷,Ñ”§±§Î,•…•Í•Î,∏þ¯Q§Î,“Á§Ï§Î,Ó䧶,÷ÿ§Õ§Î,§™§§§∑§§,∂®§·,§€
§…,…Ú§ý,§™Ó䧧,¶§È§π,◊À,ƒø§»,∑ô,õQ§·§Î,•™•Ï,’§®§Î,≤®,§´§¥,±Ø§∑§þ,“ª§ƒ,∞◊§§,“‚Œ∂
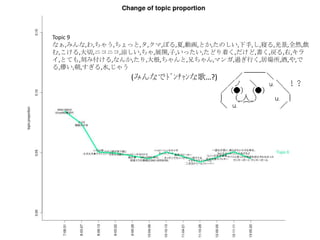
Topic 9
§ §°,§þ§Û§ ,§Ô,§¡§„§¶,§¡§Á§√§»,•ø,•Ø•Þ,§Ð§Î,œƒ,Ñ”ª≠,§»§´,§ø§Œ§∑§§,œ¬ ÷,§∑,«Þ§Î,π‚æ∞,»´»ª,Ôã§ý,§≥§±§Î,¥Û«–,•À
•≥•À•≥,õˆ§∑§§,§¡§„,’πÈ_,◊”,§§§√§ø§§,§ø§…§Í◊≈§Ø,§¿§±§…,ﯧØ,믧Î,”“,•≠•È•§,§»§∆§‚,øçþ∏∂§±§Î,§ §Û§´,§ø§Í,¥Û
∏˘,§¡§„§Û§»,–÷§¡§„§Û,•Þ•Û•¨,þ^§Æ––§Ø,æ”àˆÀ˘,æ∆,§‰,§«§Î,É觧,≥Ø,§π§Æ§Î,ÀÆ,§∏§„§¶
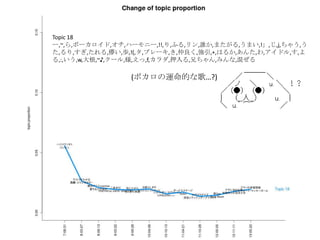
Topic 18
©`,~,§È,•Ð©`•´•Ì•§•…,•™•¡,•œ©`•‚•À©`,!!,§Í,§’§Î,•Í•Û,’l§´,§Þ§ø§¨§Î,§¶§Þ§§,!°π,§∏,j,§¡§„§¶,§¶§ø,§Î§Í,§π§Æ,§ø§Ï
§Î,É觧,öi,!(,•ø,•÷•Ï©`•≠,§≠,÷Ÿ¡º§Ø,èä“˝,+,§œ§Î§´,§¢§Û§ø,§Ô,•¢•§•…•Î,§π,§Ë§Î,:,§§§¶,w,¥Û∏˘,~?,•Ø©`•Î,òî,§®§√,f,•´
•È•¿,—∫»Î§Î,–÷§¡§„§Û,§þ§Û§ ,ªÏ§º§Î
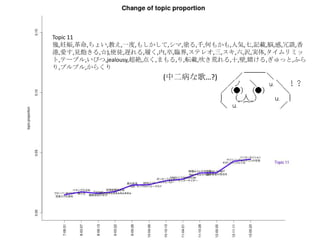
Topic 11
é◊,»—…Ô,∏Ô√¸,§¡§Á§§,Ωç®,“ª∂»,§‚§∑§´§∑§∆,•∑•Þ,âT§Î,«ß,∫Œ§‚§´§‚,»Àöð,∆þ,”õðd,√ó,∏–,»þ’Ñ,œ„∏€,ꀧπ,“äÔñ§≠
§Î,°Ó), πÕΩ,þW§Ï§Î,¬ƒ§Ø,ƒ⁄,◊‰,≈RΩÁ,•π•∆•Ï•™,»˝,•π•≠,¡˘,õg,ågÃÂ,•ø•§•ý•Í•þ•√•»,•∆©`•÷•Î,§§§”§ƒ,jealousy,≥¨Ω~,µ„
§Ø,§Þ§‚§Î,§Í,Ðûðd,¥µ§≠ªƒ§Ï§Î, Æ,±⁄,ŸÄ§±§Î,§Æ§Â§√§»,§’§È§Í,•◊•Î•◊•Î,§´§È§Ø§Í
Topic 2
ðx§Ø,...?,∏þ§§,‘™,√˧Ø,¶§È§π,ÎÖ,»’≤Ó§∑,þM§ý,§’§Ô§’§Ô,æA§Ø,≤ð‘≠,§™§∑§„§Ï,œÚ§≥§¶,∏°§Ø,•–•È,◊˜§Ï§Î,“ä…œ§≤§Î,§√
§∆§§§¶,œ£Õ˚,ƒø÷∏§π,œÞ§Í,”∞,Ωϧ±§Î,–¬§∑§§,•’•°•Û,¬Û§Ô§È√±◊”,§‚§¶§π§∞,öi§ý,Ω–§Ÿ§Î,»º§‰§π,§µ§µ§‰§´,§≠§È§≠
§È,ú∫§¡§Î,“䧃§±§Î,∫Á,∑ô,õQ§·§Î,§µ§¢,»Ð§±∫œ§¶,§‚§∞,Ó䧧,π˚§∆,Å\§ª§Î,§‰§√§»,§®,”§–§ø§Ø,§π§∞,”¬öð,¥Ú§¡ºƒ§ª§Î](https://image.slidesharecdn.com/20130609wakor2-130728054542-phpapp02/85/20130609-Wako-R-22-320.jpg)
![»’±æ’Z§«§™£Î
Topic 6
•ø,“‚Œ∂,§»§´,§´§¥,∑÷§´§Î,—®,ö𧨧ƒ§Ø,se,§ §Û§´,control,¥Û«–,∂»,øº§®§Î,§Ω§Ï§«§‚,§ø§Í,§‰,À¿§Ã,πƒÑ”,§π§Æ§Î,
§¡§„§¶,öð∑÷,§Ø§È§§,•´• ,°Ó),§∏§„§¶,¬Ñ§Ø,œ”,∫ÙŒ¸,öð§≈§Ø,§≥§π,§´§‚,•Œ,ƒÐ¡¶,þ`§¶,팧∆§Î,§∑,È]§∏§Î,Œƒ◊÷,
•þ•√•Ø•π,–—§·§Î,)°π,§¿§±§…,÷ŸÈg,§Ð§Î,Õ¥§þ,§¡§„,•≠•È•§,»þ’Ñ,è䧧,”˚§∑§§
Topic 8
Ū§®§Î,!!!!,“ªæw,ƒ–,§»§≠,ª·§¶,”◊§§,»º§®§Î,ª·§®§Î,Õ®§Íþ^§Æ§Î,Ω~åù,Àÿî≥,÷€,§µ§Ë§¶§ §È,≥Øüܧ±,Ó䧶,±Ø§∑§§,
≥ˆª·§¶,—‘§®§Î,...?,Àº§§,ÓÆ,Ö€§Ø,∂»,§Ω§Ì§Ω§Ì,þ^§Æ§Î,∞¬,±À,“䧃§·§Î,æA§Ø,ºæπù,«–§ §§,öi§ý,∞K§π,ÈvÇS,§≥§Ï
§´§È,Ωϧ±,§…§Û§ ,¶§È§π,Ó䧧,é◊§ƒ,òS§∑§§,§™§∑§„§Ï,?,¬√¡¢§ƒ,“Á§Ï§Î, À≤ð,ºs ¯,»À–Œ,?
Topic 13
§¶§Þ,Õ´,’§Ô§ª§Î,«Û§·§Î,÷ÿ§ §Î,Õ•,»Î§ÍÞz§ý,»º§‰§π,ºÁ,”≥§Î,»Ð§´§π,!!!,œ¶üܧ±,üoœÞ,ìe§Ï§Î,“䧃§·§Î,≤ð‘≠,
•“•»•Í,’Z§Í§´§±§Î,Ɉ§®§Î,•´•È,…≠,颧Î,Ω¸§≈§Ø,Î~,Ω–§÷,Ñ”§±§Î,•…•Í•Î,∏þ¯Q§Î,“Á§Ï§Î,Ó䧶,÷ÿ§Õ§Î,§™§§§∑
§§,∂®§·,§€§…,…Ú§ý,§™Ó䧧,¶§È§π,◊À,ƒø§»,∑ô,õQ§·§Î,•™•Ï,’§®§Î,≤®,§´§¥,±Ø§∑§þ,“ª§ƒ,∞◊§§,“‚Œ∂
Topic 9
§ §°,§þ§Û§ ,§Ô,§¡§„§¶,§¡§Á§√§»,•ø,•Ø•Þ,§Ð§Î,œƒ,Ñ”ª≠,§»§´,§ø§Œ§∑§§,œ¬ ÷,§∑,«Þ§Î,π‚æ∞,»´»ª,Ôã§ý,§≥§±§Î,
¥Û«–,•À•≥•À•≥,õˆ§∑§§,§¡§„,’πÈ_,◊”,§§§√§ø§§,§ø§…§Í◊≈§Ø,§¿§±§…,ﯧØ,믧Î,”“,•≠•È•§,§»§∆§‚,øçþ∏∂§±§Î,
§ §Û§´,§ø§Í,¥Û∏˘,§¡§„§Û§»,–÷§¡§„§Û,•Þ•Û•¨,þ^§Æ––§Ø,æ”àˆÀ˘,æ∆,§‰,§«§Î,É觧,≥Ø,§π§Æ§Î,ÀÆ,§∏§„§¶
Topic 18
©`,~,§È,•Ð©`•´•Ì•§•…,•™•¡,•œ©`•‚•À©`,!!,§Í,§’§Î,•Í•Û,’l§´,§Þ§ø§¨§Î,§¶§Þ§§,!°π,§∏,j,§¡§„§¶,§¶§ø,§Î§Í,§π§Æ,
§ø§Ï§Î,É觧,öi,!(,•ø,•÷•Ï©`•≠,§≠,÷Ÿ¡º§Ø,èä“˝,+,§œ§Î§´,§¢§Û§ø,§Ô,•¢•§•…•Î,§π,§Ë§Î,:,§§§¶,w,¥Û∏˘,~?,•Ø©`•Î,
òî,§®§√,f,•´•È•¿,—∫»Î§Î,–÷§¡§„§Û,§þ§Û§ ,ªÏ§º§Î
Topic 11
é◊,»—…Ô,∏Ô√¸,§¡§Á§§,Ωç®,“ª∂»,§‚§∑§´§∑§∆,•∑•Þ,âT§Î,«ß,∫Œ§‚§´§‚,»Àöð,∆þ,”õðd,√ó,∏–,»þ’Ñ,œ„∏€,ꀧπ,“äÔñ
§≠§Î,°Ó), πÕΩ,þW§Ï§Î,¬ƒ§Ø,ƒ⁄,◊‰,≈RΩÁ,•π•∆•Ï•™,»˝,•π•≠,¡˘,õg,ågÃÂ,•ø•§•ý•Í•þ•√•»,•∆©`•÷•Î,§§§”
§ƒ,jealousy,≥¨Ω~,µ„§Ø,§Þ§‚§Î,§Í,Ðûðd,¥µ§≠ªƒ§Ï§Î, Æ,±⁄,ŸÄ§±§Î,§Æ§Â§√§»,§’§È§Í,•◊•Î•◊•Î,§´§È§Ø§Í
Topic 2
ðx§Ø,...?,∏þ§§,‘™,√˧Ø,¶§È§π,ÎÖ,»’≤Ó§∑,þM§ý,§’§Ô§’§Ô,æA§Ø,≤ð‘≠,§™§∑§„§Ï,œÚ§≥§¶,∏°§Ø,•–•È,◊˜§Ï§Î,“ä…œ§≤
§Î,§√§∆§§§¶,œ£Õ˚,ƒø÷∏§π,œÞ§Í,”∞,Ωϧ±§Î,–¬§∑§§,•’•°•Û,¬Û§Ô§È√±◊”,§‚§¶§π§∞,öi§ý,Ω–§Ÿ§Î,»º§‰§π,§µ§µ§‰§´,
§≠§È§≠§È,ú∫§¡§Î,“䧃§±§Î,∫Á,∑ô,õQ§·§Î,§µ§¢,»Ð§±∫œ§¶,§‚§∞,Ó䧧,π˚§∆,Å\§ª§Î,§‰§√§»,§®,”§–§ø§Ø,§π§∞,”¬
öð,¥Ú§¡ºƒ§ª§Î](https://image.slidesharecdn.com/20130609wakor2-130728054542-phpapp02/85/20130609-Wako-R-23-320.jpg)

![Topic 6
•ø,“‚Œ∂,§»§´,§´§¥,∑÷§´§Î,—®,ö𧨧ƒ§Ø,se,§ §Û§´,control,¥Û«–,∂»,øº§®§Î,§Ω§Ï§«§‚,§ø§Í,§‰,À¿§Ã,πƒ
Ñ”,§π§Æ§Î,§¡§„§¶,öð∑÷,§Ø§È§§,•´• ,°Ó),§∏§„§¶,¬Ñ§Ø,œ”,∫ÙŒ¸,öð§≈§Ø,§≥§π,§´§‚,•Œ,ƒÐ¡¶,þ`§¶,팧∆
§Î,§∑,È]§∏§Î,Œƒ◊÷,•þ•√•Ø•π,–—§·§Î,)°π,§¿§±§…,÷ŸÈg,§Ð§Î,Õ¥§þ,§¡§„,•≠•È•§,»þ’Ñ,è䧧,”˚§∑§§
(‘™öð§ ∏Ë°≠?)](https://image.slidesharecdn.com/20130609wakor2-130728054542-phpapp02/85/20130609-Wako-R-25-320.jpg)
20130609 Wako.R •»•‘•√•Ø•‚•«•Î§Ú”√§§§ø•Ð©`•´•Ì•§•…òS«˙§Œ¡˜––Ω‚Œˆ
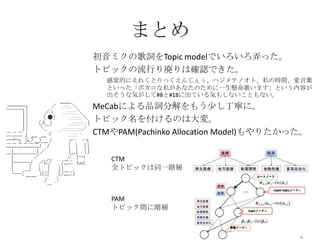
- 2. –ŒëBÀÿΩ‚Œˆ MeCab RMeCab Topic model lda, topicmodels LDA (Latent Dirichlet Allocation) DTM (Dynamic Topic Model) CTM (Correlation Topic Model) ±æ»’§ŒΩBΩÈ
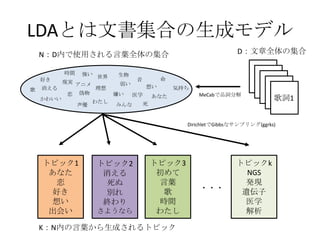
- 5. LDA§»§œŒƒï¯ºØ∫œ§Œ…˙≥…•‚•«•Î K£∫Nƒ⁄§Œ—‘»~§´§È…˙≥…§µ§Ï§Î•»•‘•√•Ø •»•‘•√•Ø1 §¢§ §ø ¡µ ∫√§≠ œÎ§§ ≥ˆª·§§ •»•‘•√•Ø3 ≥ı§·§∆ —‘»~ ∏Ë ïrÈg §Ô§ø§∑ •»•‘•√•Øk NGS ∞k¨F þzŪ◊” “Ω—ß Ω‚Œˆ ?? ? •·•Î•» ≥Ø ƒø§¨“ô§·§∆ ’ʧ√œ»§ÀÀº§§∏°§´§÷ æ˝§Œ§≥§» Àº§§«–§√§∆ «∞Ûä§Ú«–§√§ø °∏§…§¶§∑§ø§Œ£ø°π§√§∆ ¬Ñ§´§Ï§ø§Ø§∆ •‘•Û•Ø§Œ•π•´©`•» §™ª®§Œ ÛäÔó§Í §µ§∑§∆≥ˆ§´§±§Î§Œ ΩÒ»’§ŒÀΩ§œ §´§Ô§§§§§Œ§Ë£° •·•Î•» »Ð§±§∆§∑§Þ§§§Ω§¶ ∫√§≠§¿§ §Û§∆ Ω~åù§À§§§®§ §§°≠°≠ §¿§±§… •·•Î•» ƒø§‚∫œ §Ô§ª§È§Ï§ §§ ¡µ§À¡µ§ §Û§∆§∑§ §§§Ô §Ô§ø§∑ §¿§√§∆ æ˝§Œ§≥§»§¨ °≠°≠∫√§≠§ §Œ •œ•∏•·•∆•Œ•™•» ≥ı§·§∆§Œ“Ù§œ § §Û§«§∑§ø§´£ø§¢§ §ø§Œ ≥ı§·§∆§Œ“Ù§œ°≠ •Ô•ø•∑§À§»§√§∆§œ §≥§Ï§¨§Ω§¶ §¿§´§È ΩÒ §¶§Ï§∑§Ø§∆ ≥ı§·§∆§Œ—‘»~§œ § §Û§« §∑§ø§´£ø§¢§ §ø§Œ ≥ı§·§∆§Œ—‘»~ •Ô•ø•∑§œ—‘»~§√§∆ —‘§®§ §§ §¿§´§È §≥§¶§∑§∆§¶§ø§√§∆§§§Þ§π §‰§¨§∆»’§¨þ^§Æ ƒÍ§¨þ^§Æ ¿ΩÁ§¨ …´§¢ §ª§∆§‚ §¢§ §ø§¨§Ø§Ï§Î µ∆§Í§µ§®§¢§Ï§– §§§ƒ§«§‚ •Ô•ø•∑§œ§¶§ø§¶§´§È ø’§Œ…´§‚ ÔL§Œ§À§™§§§‚ ∫£§Œ…Ó§µ§‚ §¢§ §ø§Œ…˘§‚ •Ô•ø•∑§œ÷™§È § §§ §¿§±§…∏Ë§Ú ∏˧ڧ¶§ø§¶ §ø§¿…˘§Ú§¢§≤§∆ § §À§´§¢§ §ø§À ΩϧاŒ§ §È ∫Œ∂»§«§‚ ∫Œ∂»§¿§√§∆ §´§Ô§È§ §§§Ô §¢§Œ§»§≠§Œ§Þ§Þ •œ•∏•· •∆•Œ•™•»§Œ§Þ§Þ°≠ ≥ı“Ù•þ•Ø§Œœ˚ ß –≈§∏§ø§‚§Œ§œ ∂º∫œ§Œ§§§§Õ˝œÎ§Ú ¿R§Í∑µ§∑”≥§∑≥ˆ§πÁR ∏Ë䙧Ú÷π§· þµ§≠∏∂§±§Î§Ë§¶§ÀΩ–§÷ ¥Ê‘⁄“‚¡x§»§§§¶–ÈœÒ ’Ò§√§∆íB§¶§≥§»§‚§«§≠§∫ »ı §§–ƒ œ˚§®§Îø÷≤¿ «÷ ≥§π§Î±¿â≤§Ú§‚ ÷π§·§Î§€§…§Œ“‚Àº§Œè䧵 ≥ˆ¿¥§∆£®§¶§Þ§Ï£©§π§∞§Œ•Ð•Ø§œ≥÷§ø§∫ §»§∆§‚–¡§Ø±Ø§∑§Ω§¶§ Àº§§∏°§´§÷•¢• •ø §ŒÓÜ ΩK§Ô§Í§Ú∏ʧ≤ •«•£•π•◊•Ï•§§Œ÷–§«√þ§Î §≥§≥§œ§≠§√§»°∏§¥§þœ‰°π§´§ §∏§≠§À”õëõ§‚üo§Ø§ §√§∆§∑§Þ§¶§ §Û§∆ §«§‚§Õ°¢•¢• •ø§¿§±§œÕ¸ §Ï§ §§§Ë òS§∑§´§√§øïrÈg£®•»•≠£©§À øçþ∏∂§±§ø •Õ•Æ§ŒŒ∂§œ ΩÒ§‚“ô§®§∆§Î§´§ •»•‘•√•Ø1§¨∂ý§§°˙§¢§Œ»À§ÚœÎ§¶¡µ§Œ∏Ë? •»•‘•√•Ø2 œ˚§®§Î À¿§Ã Ñe§Ï ΩK§Ô§Í §µ§Ë§¶§ §È
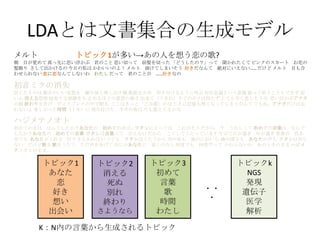
- 6. LDA§»§œŒƒï¯ºØ∫œ§Œ…˙≥…•‚•«•Î K£∫Nƒ⁄§Œ—‘»~§´§È…˙≥…§µ§Ï§Î•»•‘•√•Ø •»•‘•√•Ø1 §¢§ §ø ¡µ ∫√§≠ œÎ§§ ≥ˆª·§§ •»•‘•√•Ø3 ≥ı§·§∆ —‘»~ ∏Ë ïrÈg §Ô§ø§∑ •»•‘•√•Øk NGS ∞k¨F þzŪ◊” “Ω—ß Ω‚Œˆ ?? ? •œ•∏•·•∆•Œ•™•» ≥ı§·§∆§Œ“Ù§œ § §Û§«§∑§ø§´£ø§¢§ §ø§Œ ≥ı§·§∆§Œ“Ù§œ°≠ •Ô•ø•∑§À§»§√§∆§œ §≥§Ï§¨§Ω§¶ §¿§´§È ΩÒ §¶§Ï§∑§Ø§∆ ≥ı§·§∆§Œ—‘»~§œ § §Û§« §∑§ø§´£ø§¢§ §ø§Œ ≥ı§·§∆§Œ—‘»~ •Ô•ø•∑§œ—‘»~§√§∆ —‘§®§ §§ §¿§´§È §≥§¶§∑§∆§¶§ø§√§∆§§§Þ§π §‰§¨§∆»’§¨þ^§Æ ƒÍ§¨þ^§Æ ¿ΩÁ§¨ …´§¢ §ª§∆§‚ §¢§ §ø§¨§Ø§Ï§Î µ∆§Í§µ§®§¢§Ï§– §§§ƒ§«§‚ •Ô•ø•∑§œ§¶§ø§¶§´§È ø’§Œ…´§‚ ÔL§Œ§À§™§§§‚ ∫£§Œ…Ó§µ§‚ §¢§ §ø§Œ…˘§‚ •Ô•ø•∑§œ÷™§È § §§ §¿§±§…∏Ë§Ú ∏˧ڧ¶§ø§¶ §ø§¿…˘§Ú§¢§≤§∆ § §À§´§¢§ §ø§À ΩϧاŒ§ §È ∫Œ∂»§«§‚ ∫Œ∂»§¿§√§∆ §´§Ô§È§ §§§Ô §¢§Œ§»§≠§Œ§Þ§Þ •œ•∏•· •∆•Œ•™•»§Œ§Þ§Þ°≠ ≥ı“Ù•þ•Ø§Œœ˚ ß –≈§∏§ø§‚§Œ§œ ∂º∫œ§Œ§§§§Õ˝œÎ§Ú ¿R§Í∑µ§∑”≥§∑≥ˆ§πÁR ∏Ë䙧Ú÷π§· þµ§≠∏∂§±§Î§Ë§¶§ÀΩ–§÷ ¥Ê‘⁄“‚¡x§»§§§¶–ÈœÒ ’Ò§√§∆íB§¶§≥§»§‚§«§≠§∫ »ı §§–ƒ œ˚§®§Îø÷≤¿ «÷ ≥§π§Î±¿â≤§Ú§‚ ÷π§·§Î§€§…§Œ“‚Àº§Œè䧵 ≥ˆ¿¥§∆£®§¶§Þ§Ï£©§π§∞§Œ•Ð•Ø§œ≥÷§ø§∫ §»§∆§‚–¡§Ø±Ø§∑§Ω§¶§ Àº§§∏°§´§÷•¢• •ø §ŒÓÜ ΩK§Ô§Í§Ú∏ʧ≤ •«•£•π•◊•Ï•§§Œ÷–§«√þ§Î §≥§≥§œ§≠§√§»°∏§¥§þœ‰°π§´§ §∏§≠§À”õëõ§‚üo§Ø§ §√§∆§∑§Þ§¶§ §Û§∆ §«§‚§Õ°¢•¢• •ø§¿§±§œÕ¸ §Ï§ §§§Ë òS§∑§´§√§øïrÈg£®•»•≠£©§À øçþ∏∂§±§ø •Õ•Æ§ŒŒ∂§œ ΩÒ§‚“ô§®§∆§Î§´§ •·•Î•» ≥Ø ƒø§¨“ô§·§∆ ’ʧ√œ»§ÀÀº§§∏°§´§÷ æ˝§Œ§≥§» Àº§§«–§√§∆ «∞Ûä§Ú«–§√§ø °∏§…§¶§∑§ø§Œ£ø°π§√§∆ ¬Ñ§´§Ï§ø§Ø§∆ •‘•Û•Ø§Œ•π•´©`•» §™ª®§Œ ÛäÔó§Í §µ§∑§∆≥ˆ§´§±§Î§Œ ΩÒ»’§ŒÀΩ§œ §´§Ô§§§§§Œ§Ë£° •·•Î•» »Ð§±§∆§∑§Þ§§§Ω§¶ ∫√§≠§¿§ §Û§∆ Ω~åù§À§§§®§ §§°≠°≠ §¿§±§… •·•Î•» ƒø§‚∫œ §Ô§ª§È§Ï§ §§ ¡µ§À¡µ§ §Û§∆§∑§ §§§Ô §Ô§ø§∑ §¿§√§∆ æ˝§Œ§≥§»§¨ °≠°≠∫√§≠§ §Œ •»•‘•√•Ø1§¨∂ý§§°˙§¢§Œ»À§ÚœÎ§¶¡µ§Œ∏Ë? •»•‘•√•Ø2§¨∂ý§§°˙Ñe§Ï§Œ∏Ë? •»•‘•√•Ø2 œ˚§®§Î À¿§Ã Ñe§Ï ΩK§Ô§Í §µ§Ë§¶§ §È
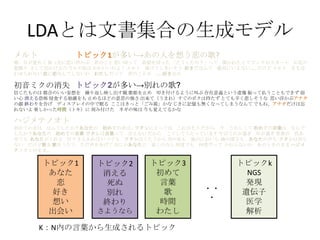
- 7. LDA§»§œŒƒï¯ºØ∫œ§Œ…˙≥…•‚•«•Î K£∫Nƒ⁄§Œ—‘»~§´§È…˙≥…§µ§Ï§Î•»•‘•√•Ø •»•‘•√•Ø1 §¢§ §ø ¡µ ∫√§≠ œÎ§§ ≥ˆª·§§ •»•‘•√•Ø3 ≥ı§·§∆ —‘»~ ∏Ë ïrÈg §Ô§ø§∑ •»•‘•√•Øk NGS ∞k¨F þzŪ◊” “Ω—ß Ω‚Œˆ ?? ? •œ•∏•·•∆•Œ•™•» ≥ı§·§∆§Œ“Ù§œ § §Û§«§∑§ø§´£ø§¢§ §ø§Œ ≥ı§·§∆§Œ“Ù§œ°≠ •Ô•ø•∑§À§»§√§∆§œ §≥§Ï§¨§Ω§¶ §¿§´§È ΩÒ §¶§Ï§∑§Ø§∆ ≥ı§·§∆§Œ—‘»~§œ § §Û§« §∑§ø§´£ø§¢§ §ø§Œ ≥ı§·§∆§Œ—‘»~ •Ô•ø•∑§œ—‘»~§√§∆ —‘§®§ §§ §¿§´§È §≥§¶§∑§∆§¶§ø§√§∆§§§Þ§π §‰§¨§∆»’§¨þ^§Æ ƒÍ§¨þ^§Æ ¿ΩÁ§¨ …´§¢ §ª§∆§‚ §¢§ §ø§¨§Ø§Ï§Î µ∆§Í§µ§®§¢§Ï§– §§§ƒ§«§‚ •Ô•ø•∑§œ§¶§ø§¶§´§È ø’§Œ…´§‚ ÔL§Œ§À§™§§§‚ ∫£§Œ…Ó§µ§‚ §¢§ §ø§Œ…˘§‚ •Ô•ø•∑§œ÷™§È § §§ §¿§±§…∏Ë§Ú ∏˧ڧ¶§ø§¶ §ø§¿…˘§Ú§¢§≤§∆ § §À§´§¢§ §ø§À ΩϧاŒ§ §È ∫Œ∂»§«§‚ ∫Œ∂»§¿§√§∆ §´§Ô§È§ §§§Ô §¢§Œ§»§≠§Œ§Þ§Þ •œ•∏•· •∆•Œ•™•»§Œ§Þ§Þ°≠ ≥ı“Ù•þ•Ø§Œœ˚ ß –≈§∏§ø§‚§Œ§œ ∂º∫œ§Œ§§§§Õ˝œÎ§Ú ¿R§Í∑µ§∑”≥§∑≥ˆ§πÁR ∏Ë䙧Ú÷π§· þµ§≠∏∂§±§Î§Ë§¶§ÀΩ–§÷ ¥Ê‘⁄“‚¡x§»§§§¶–ÈœÒ ’Ò§√§∆íB§¶§≥§»§‚§«§≠§∫ »ı §§–ƒ œ˚§®§Îø÷≤¿ «÷ ≥§π§Î±¿â≤§Ú§‚ ÷π§·§Î§€§…§Œ“‚Àº§Œè䧵 ≥ˆ¿¥§∆£®§¶§Þ§Ï£©§π§∞§Œ•Ð•Ø§œ≥÷§ø§∫ §»§∆§‚–¡§Ø±Ø§∑§Ω§¶§ Àº§§∏°§´§÷•¢• •ø §ŒÓÜ ΩK§Ô§Í§Ú∏ʧ≤ •«•£•π•◊•Ï•§§Œ÷–§«√þ§Î §≥§≥§œ§≠§√§»°∏§¥§þœ‰°π§´§ §∏§≠§À”õëõ§‚üo§Ø§ §√§∆§∑§Þ§¶§ §Û§∆ §«§‚§Õ°¢•¢• •ø§¿§±§œÕ¸ §Ï§ §§§Ë òS§∑§´§√§øïrÈg£®•»•≠£©§À øçþ∏∂§±§ø •Õ•Æ§ŒŒ∂§œ ΩÒ§‚“ô§®§∆§Î§´§ •·•Î•» ≥Ø ƒø§¨“ô§·§∆ ’ʧ√œ»§ÀÀº§§∏°§´§÷ æ˝§Œ§≥§» Àº§§«–§√§∆ «∞Ûä§Ú«–§√§ø °∏§…§¶§∑§ø§Œ£ø°π§√§∆ ¬Ñ§´§Ï§ø§Ø§∆ •‘•Û•Ø§Œ•π•´©`•» §™ª®§Œ ÛäÔó§Í §µ§∑§∆≥ˆ§´§±§Î§Œ ΩÒ»’§ŒÀΩ§œ §´§Ô§§§§§Œ§Ë£° •·•Î•» »Ð§±§∆§∑§Þ§§§Ω§¶ ∫√§≠§¿§ §Û§∆ Ω~åù§À§§§®§ §§°≠°≠ §¿§±§… •·•Î•» ƒø§‚∫œ §Ô§ª§È§Ï§ §§ ¡µ§À¡µ§ §Û§∆§∑§ §§§Ô §Ô§ø§∑ §¿§√§∆ æ˝§Œ§≥§»§¨ °≠°≠∫√§≠§ §Œ •»•‘•√•Ø1§¨∂ý§§°˙§¢§Œ»À§ÚœÎ§¶¡µ§Œ∏Ë? •»•‘•√•Ø2§¨∂ý§§°˙Ñe§Ï§Œ∏Ë? •»•‘•√•Ø3(§»•»•‘•√•Ø1)§¨∂ý§§°˙§¢§ §ø§Œ§ø§·§À≥ı§·§∆∏˧¶§Ëµƒ§ ? •»•‘•√•Ø2 œ˚§®§Î À¿§Ã Ñe§Ï ΩK§Ô§Í §µ§Ë§¶§ §È
- 8. ∂ý§Ø§Œíà’≈∞Ê
- 10. David M. Blei 2012 Dynamic Topic Model (DTM) Science’I§Œ•»•‘•√•Ø§Œâ‰þw
- 11. –°’hÕ∂∏•µ•§•»°∏–°’hº“§À§ §Ì§¶*°π§ŒΩ‚Œˆ ïrÈg≪ا»§»§‚§À Æê ¿ΩÁÐû…˙§Œ•»•‘•√•Ø§Ú∫¨§ý–°’h§¨âດ°£ •≤©`•ý§Ú“‚Œ∂§π§Î•»•‘•√•Øƒ⁄§«°¢VRMMO** §¨âດ°£ ** Virtual Reality Massively Multiplayer Online Å¢œÎ¨Fåg¥Û“郣∂ý»À ˝•™•Û•È•§•Û°£•≤©`•ýø’Èg§»§ §ÎÅ¢œÎ¨Fåg ¿ΩÁ§À •◊•Ï•§•‰©`§¨∫Œ§È§´§Œ◊∞÷√§Ú”√§§§∆ÐûÀÕ§µ§Ï°¢•«•π•≤©`•ý§∑§ø§ÍüoÀ´ §∑§ø§Í§π§Î¥Û“郣•Õ•»•≤°£ @seikichi KMC¥∫∫œÀÞ2013 * http://syosetu.com/
- 12. §…§¶øº§®§∆§‚SAO*§Œ”∞Ì맫§π°£ ±æµ±§À§¢§Í§¨§»§¶§¥§∂§§§Þ§∑§ø°£ * SWORD ART ONLINE 2022ƒÍ°¢§»§¢§Î¥Û ÷Îä◊”ôC∆˜•·©`•´©`§¨Å¢œÎø’Èg§ÿ§ŒΩ”æAôC∆˜°∂• ©`•Ù•Æ•¢°∑§ÚÈ_∞k§∑§ø§≥§»§«°¢ ¿ΩÁ§œÀϧÀÕÍ»´§ §Î•–©`•¡•„•Î•Í•¢•Í•∆•£ §Úåg¨F§µ§ª§ø°£§≥§Œ°∂• ©`•Ù•Æ•¢°∑åùèͧŒ≥ı§ŒVRMMORPG°∂•Ω©`•…•¢©`•»?•™•Û•È•§•Û£®SAO£©°∑§œ¥Û»Àö𧌃⁄§ÀÕÍ┧∑°¢Ω”æA§∑§ø1ÕÚ»À§Œ •Ê©`•∂©`§¨§Ω§Œ ¿ΩÁ§ÚòS§∑§ý§œ§∫§¿§√§ø°£§∑§´§∑°¢SAO§ÿ•¿•§•÷§∑§ø…ŸƒÍ•≠•Í•»§≥§»Õ©•ˆπ»∫Õ»À§Ú§œ§∏§·§»§∑§ø•◊•Ï•§•‰©`§ø§¡§œ°¢•≤©`•ý•Þ•π •ø©`£®GM£©§´§Èø÷§Î§Ÿ§≠”ö–˚§Ú¬Ñ§´§µ§Ï§Î°£SAO§´§È§Œ◊‘∞kµƒ•Ì•∞•¢•¶•»§œ≤ªø…ƒÐ§«§¢§Î§≥§»°¢SAO§ŒŒËî°∂∏°þ[≥«•¢•§•Û•Ø•È•√•…°∑§Œ◊Ó…œ≤ø µ⁄100唧Œ•Ð•π§Úµπ§∑§∆•≤©`•ý§Ú•Ø•Í•¢§π§Î§≥§»§¿§±§¨§≥§Œ ¿ΩÁ§´§ÈÕ—≥ˆ§π§ÎŒ®“ª§Œ∑Ω∑®§«§¢§Î§≥§»°¢§Ω§∑§∆§≥§Œ ¿ΩÁ§«À¿Õˆ§∑§øàˆ∫œ§œ¨Fåg ¿ ΩÁ§Œ•◊•Ï•§•‰©`◊‘…̧¨±æµ±§ÀÀ¿Õˆ§π§Î§»§§§¶§≥§»§Ú°£•≤©`•ý•Ø•Í•¢§Œ§ø§·§À•◊•Ï•§•‰©`§ø§¡§œÑ”§≠≥ˆ§π§¨°¢•‚•Û•π•ø©`ëȧŒî°±±§‰¨F◊¥§ÀΩ~Õ˚ §∑§ø’þ§ø§¡§À§Ë§Î◊‘ö¢§ §…§À§Ë§Í°¢È_ º§´§È1§´‘¬§«2000»À§¨À¿Õˆ°£•≤©`•ý𕬑§œ¿ßÎy§ÚòO§·§ø°£2ƒÍ··§Œ2024ƒÍ10‘¬°£°∂•¢•§•Û•Ø•È•√•…°∑§Œ ◊Ó«∞æħœµ⁄74å”°¢•◊•Ï•§•‰©`§Œ ˝§œ6000»À§€§…§À§Þ§«úp§√§∆§§§ø°£(Wiki§Œ§¢§È§π§∏) ∂˛µ∂¡˜§«üoÀ´§π§Î•≠•Í•»§µ§Û
- 13. Correlation Topic Model (CTM) LDA§œ∏˜•»•‘•√•Ø§¨∂¿¡¢§¿§»Àº§√§∆§§§Î°£ °∏ÕÍ»´§À∂¿¡¢§∑§ø•»•‘•√•Ø°π§œ¨Fågµƒ§«§œ§ §§°£ •»•‘•√•Ø§Ú¡¢§∆§π§Æ§Î§»•¿•·§À§ §Î°£ •»•‘•√•ØÈg§ŒÈvÇS§Úøºë] David M. Blei 2012
- 14. Correlation Topic Model (CTM) David M. Blei 2012
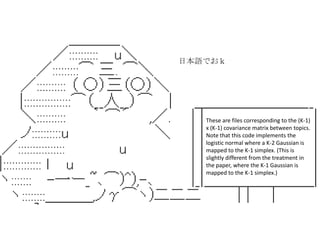
- 15. §«
- 16. topicmodels package CTM @Sigma # variance-covariance matrix of topics on the logit scale. These are files corresponding to the (K-1) x (K-1) covariance matrix between topics. Note that this code implements the logistic normal where a K-2 Gaussian is mapped to the K-1 simplex. (This is slightly different from the treatment in the paper, where the K-1 Gaussian is mapped to the K-1 simplex.) Correlation Topic Model (CTM)
- 17. These are files corresponding to the (K-1) x (K-1) covariance matrix between topics. Note that this code implements the logistic normal where a K-2 Gaussian is mapped to the K-1 simplex. (This is slightly different from the treatment in the paper, where the K-1 Gaussian is mapped to the K-1 simplex.) »’±æ’Z§«§™£Î
- 18. (K-1) x (K-1) covariance matrix?? •»•‘•√•Ø ˝§œK§¿§Ì? Note that this code implements the logistic normal where a K-2 Gaussian is mapped to the K-1 simplex. La estoy cr®™ptica distribuci®Æn de Gauss de K-2 ?Qui®¶n dijo que corresponde a la simple de K-1? (K-2§Œ•¨•¶•π∑÷≤º§¨K-1§Œsimplex§Àåùèͧ∑§∆§§§Î§√§∆ “‚Œ∂≤ª√˜§ §Û§¿§±§…?) ???
- 20. Dynamic Topic Model (DTM) on Vocaloid musics LDA§¿§±§¿§»§ƒ§Þ§È§ §§°£ CTM§œ≥ˆ¡¶§¨“‚Œ∂≤ª√˜°£ °∏–°’hº“§À§ §Ì§¶°π§Œ∂˛∑¨ºÂ§∏§¿§¨DTM§Ú§‰§Ì§¶ ≥ı“Ù•þ•Ø Wiki (∏Ë‘~÷√àˆ)§´§È∏Ë‘~§Ú•π•Ø•Ï•§•‘•Û•∞°£ DTM§œR§À§ §§!?!? C++§Œ•È•§•÷•È•Í©`§Ú¿˚”√§∑§ø°£ •—•È•·©`•ø •»•‘•√•Ø ˝ K=30
- 22. Topic 6 •ø,“‚Œ∂,§»§´,§´§¥,∑÷§´§Î,—®,ö𧨧ƒ§Ø,se,§ §Û§´,control,¥Û«–,∂»,øº§®§Î,§Ω§Ï§«§‚,§ø§Í,§‰,À¿§Ã,πƒÑ”,§π§Æ§Î,§¡§„ §¶,öð∑÷,§Ø§È§§,•´• ,°Ó),§∏§„§¶,¬Ñ§Ø,œ”,∫ÙŒ¸,öð§≈§Ø,§≥§π,§´§‚,•Œ,ƒÐ¡¶,þ`§¶,팧∆§Î,§∑,È]§∏§Î,Œƒ◊÷,•þ•√•Ø•π,–—§· §Î,)°π,§¿§±§…,÷ŸÈg,§Ð§Î,Õ¥§þ,§¡§„,•≠•È•§,»þ’Ñ,è䧧,”˚§∑§§ Topic 8 Ū§®§Î,!!!!,“ªæw,ƒ–,§»§≠,ª·§¶,”◊§§,»º§®§Î,ª·§®§Î,Õ®§Íþ^§Æ§Î,Ω~åù,Àÿî≥,÷€,§µ§Ë§¶§ §È,≥Øüܧ±,Ó䧶,±Ø§∑§§,≥ˆª· §¶,—‘§®§Î,...?,Àº§§,ÓÆ,Ö€§Ø,∂»,§Ω§Ì§Ω§Ì,þ^§Æ§Î,∞¬,±À,“䧃§·§Î,æA§Ø,ºæπù,«–§ §§,öi§ý,∞K§π,ÈvÇS,§≥§Ï§´§È,Ωϧ±,§… §Û§ ,¶§È§π,Ó䧧,é◊§ƒ,òS§∑§§,§™§∑§„§Ï,?,¬√¡¢§ƒ,“Á§Ï§Î, À≤ð,ºs ¯,»À–Œ,? Topic 13 §¶§Þ,Õ´,’§Ô§ª§Î,«Û§·§Î,÷ÿ§ §Î,Õ•,»Î§ÍÞz§ý,»º§‰§π,ºÁ,”≥§Î,»Ð§´§π,!!!,œ¶üܧ±,üoœÞ,ìe§Ï§Î,“䧃§·§Î,≤ð‘≠,•“•» •Í,’Z§Í§´§±§Î,Ɉ§®§Î,•´•È,…≠,颧Î,Ω¸§≈§Ø,Î~,Ω–§÷,Ñ”§±§Î,•…•Í•Î,∏þ¯Q§Î,“Á§Ï§Î,Ó䧶,÷ÿ§Õ§Î,§™§§§∑§§,∂®§·,§€ §…,…Ú§ý,§™Ó䧧,¶§È§π,◊À,ƒø§»,∑ô,õQ§·§Î,•™•Ï,’§®§Î,≤®,§´§¥,±Ø§∑§þ,“ª§ƒ,∞◊§§,“‚Œ∂ Topic 9 § §°,§þ§Û§ ,§Ô,§¡§„§¶,§¡§Á§√§»,•ø,•Ø•Þ,§Ð§Î,œƒ,Ñ”ª≠,§»§´,§ø§Œ§∑§§,œ¬ ÷,§∑,«Þ§Î,π‚æ∞,»´»ª,Ôã§ý,§≥§±§Î,¥Û«–,•À •≥•À•≥,õˆ§∑§§,§¡§„,’πÈ_,◊”,§§§√§ø§§,§ø§…§Í◊≈§Ø,§¿§±§…,ﯧØ,믧Î,”“,•≠•È•§,§»§∆§‚,øçþ∏∂§±§Î,§ §Û§´,§ø§Í,¥Û ∏˘,§¡§„§Û§»,–÷§¡§„§Û,•Þ•Û•¨,þ^§Æ––§Ø,æ”àˆÀ˘,æ∆,§‰,§«§Î,É觧,≥Ø,§π§Æ§Î,ÀÆ,§∏§„§¶ Topic 18 ©`,~,§È,•Ð©`•´•Ì•§•…,•™•¡,•œ©`•‚•À©`,!!,§Í,§’§Î,•Í•Û,’l§´,§Þ§ø§¨§Î,§¶§Þ§§,!°π,§∏,j,§¡§„§¶,§¶§ø,§Î§Í,§π§Æ,§ø§Ï §Î,É觧,öi,!(,•ø,•÷•Ï©`•≠,§≠,÷Ÿ¡º§Ø,èä“˝,+,§œ§Î§´,§¢§Û§ø,§Ô,•¢•§•…•Î,§π,§Ë§Î,:,§§§¶,w,¥Û∏˘,~?,•Ø©`•Î,òî,§®§√,f,•´ •È•¿,—∫»Î§Î,–÷§¡§„§Û,§þ§Û§ ,ªÏ§º§Î Topic 11 é◊,»—…Ô,∏Ô√¸,§¡§Á§§,Ωç®,“ª∂»,§‚§∑§´§∑§∆,•∑•Þ,âT§Î,«ß,∫Œ§‚§´§‚,»Àöð,∆þ,”õðd,√ó,∏–,»þ’Ñ,œ„∏€,ꀧπ,“äÔñ§≠ §Î,°Ó), πÕΩ,þW§Ï§Î,¬ƒ§Ø,ƒ⁄,◊‰,≈RΩÁ,•π•∆•Ï•™,»˝,•π•≠,¡˘,õg,ågÃÂ,•ø•§•ý•Í•þ•√•»,•∆©`•÷•Î,§§§”§ƒ,jealousy,≥¨Ω~,µ„ §Ø,§Þ§‚§Î,§Í,Ðûðd,¥µ§≠ªƒ§Ï§Î, Æ,±⁄,ŸÄ§±§Î,§Æ§Â§√§»,§’§È§Í,•◊•Î•◊•Î,§´§È§Ø§Í Topic 2 ðx§Ø,...?,∏þ§§,‘™,√˧Ø,¶§È§π,ÎÖ,»’≤Ó§∑,þM§ý,§’§Ô§’§Ô,æA§Ø,≤ð‘≠,§™§∑§„§Ï,œÚ§≥§¶,∏°§Ø,•–•È,◊˜§Ï§Î,“ä…œ§≤§Î,§√ §∆§§§¶,œ£Õ˚,ƒø÷∏§π,œÞ§Í,”∞,Ωϧ±§Î,–¬§∑§§,•’•°•Û,¬Û§Ô§È√±◊”,§‚§¶§π§∞,öi§ý,Ω–§Ÿ§Î,»º§‰§π,§µ§µ§‰§´,§≠§È§≠ §È,ú∫§¡§Î,“䧃§±§Î,∫Á,∑ô,õQ§·§Î,§µ§¢,»Ð§±∫œ§¶,§‚§∞,Ó䧧,π˚§∆,Å\§ª§Î,§‰§√§»,§®,”§–§ø§Ø,§π§∞,”¬öð,¥Ú§¡ºƒ§ª§Î
- 23. »’±æ’Z§«§™£Î Topic 6 •ø,“‚Œ∂,§»§´,§´§¥,∑÷§´§Î,—®,ö𧨧ƒ§Ø,se,§ §Û§´,control,¥Û«–,∂»,øº§®§Î,§Ω§Ï§«§‚,§ø§Í,§‰,À¿§Ã,πƒÑ”,§π§Æ§Î, §¡§„§¶,öð∑÷,§Ø§È§§,•´• ,°Ó),§∏§„§¶,¬Ñ§Ø,œ”,∫ÙŒ¸,öð§≈§Ø,§≥§π,§´§‚,•Œ,ƒÐ¡¶,þ`§¶,팧∆§Î,§∑,È]§∏§Î,Œƒ◊÷, •þ•√•Ø•π,–—§·§Î,)°π,§¿§±§…,÷ŸÈg,§Ð§Î,Õ¥§þ,§¡§„,•≠•È•§,»þ’Ñ,è䧧,”˚§∑§§ Topic 8 Ū§®§Î,!!!!,“ªæw,ƒ–,§»§≠,ª·§¶,”◊§§,»º§®§Î,ª·§®§Î,Õ®§Íþ^§Æ§Î,Ω~åù,Àÿî≥,÷€,§µ§Ë§¶§ §È,≥Øüܧ±,Ó䧶,±Ø§∑§§, ≥ˆª·§¶,—‘§®§Î,...?,Àº§§,ÓÆ,Ö€§Ø,∂»,§Ω§Ì§Ω§Ì,þ^§Æ§Î,∞¬,±À,“䧃§·§Î,æA§Ø,ºæπù,«–§ §§,öi§ý,∞K§π,ÈvÇS,§≥§Ï §´§È,Ωϧ±,§…§Û§ ,¶§È§π,Ó䧧,é◊§ƒ,òS§∑§§,§™§∑§„§Ï,?,¬√¡¢§ƒ,“Á§Ï§Î, À≤ð,ºs ¯,»À–Œ,? Topic 13 §¶§Þ,Õ´,’§Ô§ª§Î,«Û§·§Î,÷ÿ§ §Î,Õ•,»Î§ÍÞz§ý,»º§‰§π,ºÁ,”≥§Î,»Ð§´§π,!!!,œ¶üܧ±,üoœÞ,ìe§Ï§Î,“䧃§·§Î,≤ð‘≠, •“•»•Í,’Z§Í§´§±§Î,Ɉ§®§Î,•´•È,…≠,颧Î,Ω¸§≈§Ø,Î~,Ω–§÷,Ñ”§±§Î,•…•Í•Î,∏þ¯Q§Î,“Á§Ï§Î,Ó䧶,÷ÿ§Õ§Î,§™§§§∑ §§,∂®§·,§€§…,…Ú§ý,§™Ó䧧,¶§È§π,◊À,ƒø§»,∑ô,õQ§·§Î,•™•Ï,’§®§Î,≤®,§´§¥,±Ø§∑§þ,“ª§ƒ,∞◊§§,“‚Œ∂ Topic 9 § §°,§þ§Û§ ,§Ô,§¡§„§¶,§¡§Á§√§»,•ø,•Ø•Þ,§Ð§Î,œƒ,Ñ”ª≠,§»§´,§ø§Œ§∑§§,œ¬ ÷,§∑,«Þ§Î,π‚æ∞,»´»ª,Ôã§ý,§≥§±§Î, ¥Û«–,•À•≥•À•≥,õˆ§∑§§,§¡§„,’πÈ_,◊”,§§§√§ø§§,§ø§…§Í◊≈§Ø,§¿§±§…,ﯧØ,믧Î,”“,•≠•È•§,§»§∆§‚,øçþ∏∂§±§Î, § §Û§´,§ø§Í,¥Û∏˘,§¡§„§Û§»,–÷§¡§„§Û,•Þ•Û•¨,þ^§Æ––§Ø,æ”àˆÀ˘,æ∆,§‰,§«§Î,É觧,≥Ø,§π§Æ§Î,ÀÆ,§∏§„§¶ Topic 18 ©`,~,§È,•Ð©`•´•Ì•§•…,•™•¡,•œ©`•‚•À©`,!!,§Í,§’§Î,•Í•Û,’l§´,§Þ§ø§¨§Î,§¶§Þ§§,!°π,§∏,j,§¡§„§¶,§¶§ø,§Î§Í,§π§Æ, §ø§Ï§Î,É觧,öi,!(,•ø,•÷•Ï©`•≠,§≠,÷Ÿ¡º§Ø,èä“˝,+,§œ§Î§´,§¢§Û§ø,§Ô,•¢•§•…•Î,§π,§Ë§Î,:,§§§¶,w,¥Û∏˘,~?,•Ø©`•Î, òî,§®§√,f,•´•È•¿,—∫»Î§Î,–÷§¡§„§Û,§þ§Û§ ,ªÏ§º§Î Topic 11 é◊,»—…Ô,∏Ô√¸,§¡§Á§§,Ωç®,“ª∂»,§‚§∑§´§∑§∆,•∑•Þ,âT§Î,«ß,∫Œ§‚§´§‚,»Àöð,∆þ,”õðd,√ó,∏–,»þ’Ñ,œ„∏€,ꀧπ,“äÔñ §≠§Î,°Ó), πÕΩ,þW§Ï§Î,¬ƒ§Ø,ƒ⁄,◊‰,≈RΩÁ,•π•∆•Ï•™,»˝,•π•≠,¡˘,õg,ågÃÂ,•ø•§•ý•Í•þ•√•»,•∆©`•÷•Î,§§§” §ƒ,jealousy,≥¨Ω~,µ„§Ø,§Þ§‚§Î,§Í,Ðûðd,¥µ§≠ªƒ§Ï§Î, Æ,±⁄,ŸÄ§±§Î,§Æ§Â§√§»,§’§È§Í,•◊•Î•◊•Î,§´§È§Ø§Í Topic 2 ðx§Ø,...?,∏þ§§,‘™,√˧Ø,¶§È§π,ÎÖ,»’≤Ó§∑,þM§ý,§’§Ô§’§Ô,æA§Ø,≤ð‘≠,§™§∑§„§Ï,œÚ§≥§¶,∏°§Ø,•–•È,◊˜§Ï§Î,“ä…œ§≤ §Î,§√§∆§§§¶,œ£Õ˚,ƒø÷∏§π,œÞ§Í,”∞,Ωϧ±§Î,–¬§∑§§,•’•°•Û,¬Û§Ô§È√±◊”,§‚§¶§π§∞,öi§ý,Ω–§Ÿ§Î,»º§‰§π,§µ§µ§‰§´, §≠§È§≠§È,ú∫§¡§Î,“䧃§±§Î,∫Á,∑ô,õQ§·§Î,§µ§¢,»Ð§±∫œ§¶,§‚§∞,Ó䧧,π˚§∆,Å\§ª§Î,§‰§√§»,§®,”§–§ø§Ø,§π§∞,”¬ öð,¥Ú§¡ºƒ§ª§Î
- 33. §Þ§»§· ≥ı“Ù•þ•Ø§Œ∏Ë‘~§ÚTopic model§«§§§Ì§§§Ì≈™§√§ø°£ •»•‘•√•Ø§Œ¡˜––§Í鸧ͧœ¥_’J§«§≠§ø°£ ∏–“ôµƒ§À§®§Ï§Ø§»§Í§√§Ø§®§Û§∏§ß§•°¢•œ•∏•·•∆•Œ•™•»°¢ÀΩ§ŒïrÈg°¢ê€—‘»~ §»§§§√§ø°∏•Ð•´•Ì§ ÀΩ§¨§¢§ §ø§Œ§ø§·§À“ª…˙ë“√¸∏˧§§Þ§π°π§»§§§¶ƒ⁄»ð§¨ ≥ˆ§Ω§¶§ ö𧨧∑§∆#8§»#18§À≥ˆ§∆§§§Îö𧂧∑§ §§§≥§»§‚§ §§°£ MeCab§À§Ë§Î∆∑‘~∑÷Ω‚§Ú§‚§¶…Ÿ§∑∂°åé§À°£ •»•‘•√•Ø√˚§Ú∏∂§±§Î§Œ§œ¥Û≰£ CTM§‰PAM(Pachinko Allocation Model)§‚§‰§Í§ø§´§√§ø°£ CTM »´•»•‘•√•Ø§œÕ¨“ªÎAå” PAM •»•‘•√•ØÈg§ÀÎAå”