






































More Related Content
Similar to Sequence Alignment - Data Bioinformatics Introduction (20)
Recently uploaded (20)


















Sequence Alignment - Data Bioinformatics Introduction
- 1. Learning from primary structure Sequence alignment
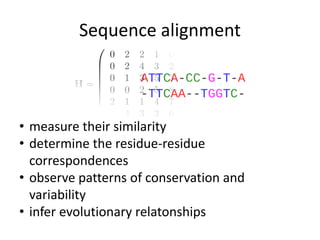
- 2. Sequence alignment • measure their similarity • determine the residue-residue correspondences • observe patterns of conservation and variability • infer evolutionary relatonships
- 3. Measure of similarity alignment: identification of residue-residue correspondences Correspondences must preserve the order of residues Gaps may be introduced Example: First string= a b c d e second string= a c d e f A reasonable alignment: a b c d e – a – c d e f
- 4. Measure of similarity We must define criteria so that an algorithm can choose the best alignment Example: gctgaacg ctataatc Alignments: - - - - - - - g c t g a a c g c t a t a a t c - - - - - - - g c t g a a c g c t a t a a t c g c t g a - a - - c g - - c t - a t a a t c g c t g - a a - c g - c t a t a a t c -
- 5. Measure of similarity We need a way to examine all possible alignments systematically. Then we need to compute a score reflecting the quality of each possible alignment, and to identify the alignment with the optimal score Several different alignments may give the same best score Even minor variations in the scoring scheme may change the ranking of alignments, causing a different one to emerge as the best
- 6. Dotplot • give an overview of the similarities between two sequences • have a close relationship with the alignment between two sequences Da: Lesk, Introduction to Bioinformatics Dotplot showing identities between short name (DOROTHYHODGKIN) and full name (DOROTHYCROWFOOTH ODGKIN)
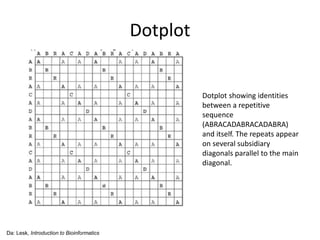
- 7. Dotplot Da: Lesk, Introduction to Bioinformatics Dotplot showing identities between a repetitive sequence (ABRACADABRACADABRA) and itself. The repeats appear on several subsidiary diagonals parallel to the main diagonal.
- 8. Dotplot Da: Lesk, Introduction to Bioinformatics Dotplot showing identities between the palindromic sequence MAX I STAY AWAY AT SIX AM and itself. The palindrome reveals itself as a stretch of matches perpendicular to the main diagonal Remember that: Restriction enzymes and transcriptional regulatory factors may recognize palindrome sequences EcoRI: GAATTC CTTAAG
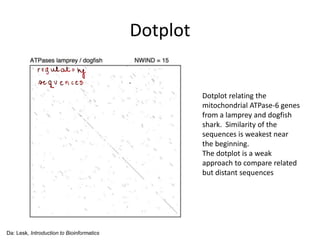
- 9. Dotplot Da: Lesk, Introduction to Bioinformatics Dotplot relating the mitochondrial ATPase-6 genes from a lamprey and dogfish shark. Similarity of the sequences is weakest near the beginning. The dotplot is a weak approach to compare related but distant sequences
- 10. Dotplot Proteins dotplot: a dotplot relating PAX-6 protein of mouse and the eyeless protein of Drosophila melanogaster. The mouse sequence shows an insertion that is missing in Drosophila Rielaborato da: Lesk, Introduction to Bioinformatics
- 11. Dotplot and sequence alignment The dotplot capture the overall similarity of two sequences and also the complete set and relative quality of different possible alignments. Diagonal movement indicates that the residues align; horizontal movement indicates that a gap must be introduced in the sequence shown in the lines; if it is vertical, the gap is introduced in the column sequence Da: Lesk, Introduction to Bioinformatics DOROTHY--------HODGKIN DOROTHYCROWFOOTHODGKIN
- 12. Measures of sequence similarity Given two character strings, two measures of the distance between them are: • The Hamming distance, defined between two strings of equal length, is the number of positions with mismatching characters. • The Levenshtein, or edit distance, between two strigs of not necessarily equal length, is the minimal number of ’edit operations’ required to change one string into the other, where an edit operation is a deletion, insertion or alteration of a single chracter in either sequence. For example: agtc Hamming distance = 2 cgta ag-tcc Levenshtein distance = 3 cgctca Da: Lesk, Introduction to Bioinformatics