
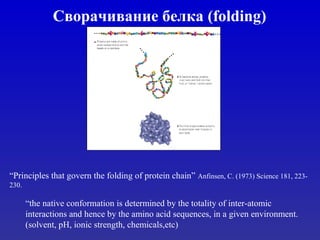












![1. ąÆąĄčĆąŠčÅčéąĮąŠčüčéąĖ
P╬▒(H)=[(#H in helix)/(#H)]/(fraction helix {all})
T S P T A E L M R S T G
P(H) 69 77 57 69 142 151 121 145 98 77 69 57
P(E) 147 75 55 147 83 37 130 105 93 75 147 75
P(turn) 114 143 152 114 66 74 59 60 95 143 114 156
ąĀą░ąĘą▓ąĖčéąĖąĄ Chou-Fasman](https://image.slidesharecdn.com/vvedenievbioinformatiku4-130814090448-phpapp01/85/Vvedenie-v-bioinformatiku_4-14-320.jpg)





































































More Related Content
Similar to Vvedenie v bioinformatiku_4 (20)
More from BioinformaticsInstitute (20)


















Vvedenie v bioinformatiku_4
- 1. ąĪą▓ąŠčĆą░čćąĖą▓ą░ąĮąĖąĄ ą▒ąĄą╗ą║ą░ (folding) ŌĆ£Principles that govern the folding of protein chainŌĆØ Anfinsen, C. (1973) Science 181, 223- 230. ŌĆ£the native conformation is determined by the totality of inter-atomic interactions and hence by the amino acid sequences, in a given environment. (solvent, pH, ionic strength, chemicals,etc)
- 2. ą¤čĆąŠą▒ą╗ąĄą╝ą░ čüą▓ąŠčĆą░čćąĖą▓ą░ąĮąĖčÅ ą▒ąĄą╗ą║ą░ ŌĆóążčāąĮą║čåąĖąĖ ą▒ąĄą╗ą║ąŠą▓ ąŠą┐čĆąĄą┤ąĄą╗čÅčÄčéčüčÅ ąĖčüą║ą╗čÄčćąĖč鹥ą╗čīąĮąŠ ąĖčģ 3D čüčéčĆčāą║čéčāčĆąŠą╣, ą║ąŠąĮč乊čĆą╝ą░čåąĖąĄą╣ ŌĆóąÆąŠą┐čĆąŠčü: ą╝ąŠąČąĮąŠ ą╗ąĖ ą┐čĆąĄą┤čüą║ą░ąĘą░čéčī 3D čüčéčĆčāą║čéčāčĆčā ą▒ąĄą╗ą║ą░, ąĖčüčģąŠą┤čÅ ąĖąĘ ą┤ą░ąĮąĮąŠą╣ ą░ą╝ąĖąĮąŠą║ąĖčüą╗ąŠčéąĮąŠą╣ ą┐ąŠčüą╗ąĄą┤ąŠą▓ą░č鹥ą╗čīąĮąŠčüčéąĖ? ŌĆóą×čéą▓ąĄčé: ą▓ ąŠą▒čēąĄą╝ čüą╗čāčćą░ąĄ ŌĆō ąĮąĄčé! ŌĆó ąØąŠ ą╝ąĄč鹊ą┤čŗ, ą║ąŠč鹊čĆčŗąĄ ą┐ąŠąĘą▓ąŠą╗čÅčÄčé čĆą░ąĘčĆąĄčłąĖčéčī čćą░čüčéąĖčćąĮąŠ 3D čüčéčĆčāą║čéčāčĆčŗ, ą▒čŗą▓ą░čÄčé ą┐ąŠą╗ąĄąĘąĮčŗ.
- 3. ąĪą▓ąŠčĆą░čćąĖą▓ą░ąĮąĖąĄ ŌĆō ą┐ąŠč湥ą╝čā čŹč鹊 čéą░ą║ čüą╗ąŠąČąĮąŠ? ŌĆó ąøąĖąĮąĄą╣ąĮčŗąĄ ą╝ąŠą╗ąĄą║čāą╗čŗ ą▒ąĄą╗ą║ąŠą▓ ąŠč湥ąĮčī ą▒čŗčüčéčĆąŠ čüą▓ąŠčĆą░čćąĖą▓ą░čÄčéčüčÅ ą▓ ą┐čĆąĄą┤ąŠą┐čĆąĄą┤ąĄą╗čæąĮąĮčŗąĄ 3D čüčéčĆčāą║čéčāčĆčŗ. ŌĆó ąĪą▓ąŠą╣čüčéą▓ą░ ą╗čÄą▒ąŠą│ąŠ ą▒ąĄą╗ą║ą░ ąŠą┐čĆąĄą┤ąĄą╗čÅčÄčéčüčÅ ąĄą│ąŠ 3D čüčéčĆčāą║čéčāčĆąŠą╣. ŌĆó ąæąĄą╗ą║ąĖ ą╝ąŠą│čāčé ą┤ąĄąĮą░čéčāčĆąĖčĆąŠą▓ą░čéčī ą┐ąŠą┤ ą▓ąŠąĘą┤ąĄą╣čüčéą▓ąĖąĄą╝ čģąĖą╝ąĖč湥čüą║ąĖčģ ą▓ąĄčēąĄčüčéą▓ ąĖą╗ąĖ č鹥ą┐ą╗ąŠčéčŗ, ąĮąŠ ąĘą░č鹥ą╝ ąŠąĮąĖ čüą▓ąŠčĆą░čćąĖą▓ą░čÄčéčüčÅ ą▓ąĮąŠą▓čī ą▓ ąĖčüčģąŠą┤ąĮčāčÄ čüčéčĆčāą║čéčāčĆčā. ąóą░ą║ ą┐ąŠč湥ą╝čā ąČąĄ čéą░ą║ čüą╗ąŠąČąĮąŠ čĆą░ąĘčĆąĄčłąĖčéčī čüą▓ąŠčĆą░čćąĖą▓ą░ąĮąĖąĄ? ŌĆó ąĪčéčĆčāą║čéčāčĆą░ ą▒ąĄą╗ą║ą░ ą╝ąŠąČąĄčé ą▒čŗčéčī ąŠą┐čĆąĄą┤ąĄą╗ąĄąĮą░ 菹║čüą┐ąĄčĆąĖą╝ąĄąĮčéą░ą╗čīąĮąŠ (X-Rays or NMR) ąĮąŠ čŹčéą░ ą┐čĆąŠčåąĄą┤čāčĆą░ ąĮąĄ ą▓čüąĄą│ą┤ą░ ą▓ąŠąĘą╝ąŠąČąĮą░ ąĖ ąĮąĄ ą▓čüąĄą│ą┤ą░ ą┤ą░čæčé čģąŠčĆąŠčłąĖąĄ čĆąĄąĘčāą╗čīčéą░čéčŗ. ŌĆó 3D čüčéčĆčāą║čéčāčĆą░ ą┐ąŠčüą╗ąĄą┤ąŠą▓ą░č鹥ą╗čīąĮąŠčüčéąĖ ąŠą┐čĆąĄą┤ąĄą╗čÅąĄčéčüčÅ ą┐ąŠčüą╗ąĄą┤ąŠą▓ą░č鹥ą╗čīąĮąŠčüčéčīčÄ ą┤ąŠą┐čāčüčéąĖą╝čŗčģ čāą│ą╗ąŠą▓ ą┐ąŠą▓ąŠčĆąŠčéą░, ą▓ ą║ąŠč鹊čĆąŠą╣ ą║ą░ąČą┤čŗą╣ čāą│ąŠą╗ ┬½čüąŠčüč鹊ąĖčé┬╗ ąĖąĘ 2-čģ ą┐ą╗ą░ąĮą░čĆąĮčŗčģ čāą│ą╗ąŠą▓. ŌĆó ąŁčéą░ ą┐čĆąŠą▒ą╗ąĄą╝ą░ ą╝ąŠąČąĄčé ą▒čŗčéčī čĆąĄčłąĄąĮą░ ą┐čāčéčæą╝ ą┤ąĖčüą║čĆąĄčéąĖąĘą░čåąĖąĖ (ą┐čĆąĖ ąĮąĄą║ąŠč鹊čĆąŠą╣ ą┐ąŠč鹥čĆąĄ č鹊čćąĮąŠčüčéąĖ) ą┐čāčéčæą╝ ąŠą│čĆą░ąĮąĖč湥ąĮąĖčÅ ą║ąŠą╗ąĖč湥čüčéą▓ą░ ą▓ąŠąĘą╝ąŠąČąĮčŗčģ ą┐čāč鹥ą╣ ą┤ąŠčüčéąĖąČąĄąĮąĖčÅ ą║ą░ąČą┤ąŠą╣ ąĖąĘ č鹊č湥ą║ (ą║ąŠąŠčĆą┤ąĖąĮą░čé ą░č鹊ą╝ąŠą▓).
- 4. ąĢčüą╗ąĖ čā ą▓ą░čü ąĮąĄčé čüčéčĆčāą║čéčāčĆčŗŌĆ”
- 5. ą¤ąŠą┤čģąŠą┤čŗ ą▓ ą┐čĆąĄą┤čüą║ą░ąĘą░ąĮąĖąĖ čüčéčĆčāą║čéčāčĆčŗ ŌĆó ą¤čĆąĄą┤čüą║ą░ąĘą░ąĮąĖąĄ 1D: ŌĆō ą▓č鹊čĆąĖčćąĮčŗąĄ čüčéčĆčāą║čéčāčĆčŗ ŌĆōą┤ąŠčüčéčāą┐ąĮąŠčüčéčī ą┤ą╗čÅ čĆą░čüčéą▓ąŠčĆąĖč鹥ą╗čÅ ŌĆō čéčĆą░ąĮčüą╝ąĄą╝ą▒čĆą░ąĮą░ą╗čīąĮčŗąĄ čüą┐ąĖčĆą░ą╗ąĖ ŌĆó ą¤čĆąĄą┤čüą║ą░ąĮąĖąĄ 2D: ŌĆō ą║ąŠąĮčéą░ą║čéčŗ ą╝ąĄąČą┤čā ą░ą╝ąĖąĮąŠą║ąĖčüą╗ąŠčéą░ą╝ąĖ/ąĮąĖčéčÅą╝ąĖ ą▒ąĄčéą░. ŌĆó ą¤čĆąĄą┤čüą║ą░ąĘą░ąĮąĖąĄ 3D: ŌĆōą╝ąŠą┤ąĄą╗ąĖčĆąŠą▓ą░ąĮąĖąĄ ą│ąŠą╝ąŠą╗ąŠą│ąŠą▓ ŌĆō čĆą░čüą┐ąŠąĘąĮą░ą▓ą░ąĮąĖąĄ č乊ą╗ą┤ą░ (e.g. via threading) ŌĆō ab initio ą┐čĆąĄą┤čüą║ą░ąĘą░ąĮąĖąĄ (e.g. via ą╝ąŠą╗ąĄą║čāą╗čÅčĆąĮą░čÅ ą┤ąĖąĮą░ą╝ąĖą║ą░)
- 6. ąŚą░ą┤ą░čćąĖ ŌĆó ąĪčĆą░ą▓ąĮąĄąĮąĖąĄ ą▓čüąĄčģ ąĖąĘą▓ąĄčüčéąĮčŗčģ čüčéčĆčāą║čéčāčĆ ą┤čĆčāą│ čü ą┤čĆčāą│ąŠą╝ ŌĆó ąÜą╗ą░čüčüąĖčäąĖą║ą░čåąĖčÅ ąĖ ąŠčĆą│ą░ąĮąĖąĘą░čåąĖčÅ ą▓čüąĄčģ ąĖąĘą▓ąĄčüčéąĮčŗčģ čüčéčĆčāą║čéčāčĆ ŌĆó ą¤ąŠąĖčüą║ ąŠą▒čēąĖčģ čüčéčĆčāą║čéčāčĆąĮčŗčģ čłą░ą▒ą╗ąŠąĮąŠą▓ ąĖ ą╝ąŠčéąĖą▓ąŠą▓ ŌĆó ą×ą┐čĆąĄą┤ąĄą╗ąĄąĮąĖąĄ 菹▓ąŠą╗čÄčåąĖąŠąĮąĮčŗčģ čĆą░čüčüč鹊čÅąĮąĖą╣ ą╝ąĄąČą┤čā čüčéčĆčāą║čéčāčĆą░ą╝ąĖ ą▒ąĄą╗ą║ąŠą▓ ŌĆó ąöąŠą║ąĖąĮą│ ŌĆō ąĖąĘčāč湥ąĮąĖąĄ ą▓ąĘą░ąĖą╝ąŠą┤ąĄą╣čüčéą▓ąĖčÅ ą╝ąĄąČą┤čā čüčéčĆčāą║čéčāčĆą░ą╝ąĖ ŌĆó ą¤čĆąĄą┤čüą║ą░ąĘą░ąĮąĖąĄ čüčéčĆčāą║čéčāčĆ ąĮą░ ąŠčüąĮąŠą▓ąĄ ą┐ąŠčüą╗ąĄą┤ąŠą▓ą░č鹥ą╗čīąĮąŠčüčéąĖ ŌĆó ąöąĖąĘą░ą╣ąĮ ąĮąŠą▓čŗčģ ą╗ąĄą║ą░čĆčüčéą▓
- 7. ąŚą░č湥ą╝? ŌĆó ą¤ąĄčĆą▓čŗą╣ čłą░ą│ ą║ ą┐čĆąĄą┤čüą║ą░ąĘą░ąĮąĖčÄ čéčĆąĄčéąĖčćąĮąŠą╣ čüčéčĆčāą║čéčāčĆčŗ ŌĆó ą×ą┤ąĖąĮ ąĖąĘ ąŠčüąĮąŠą▓ąĮčŗčģ 菹╗ąĄą╝ąĄąĮč鹊ą▓ ą▓ čĆą░čüą┐ąŠąĘąĮą░ą▓ą░ąĮąĖąĖ č乊ą╗ą┤ą░ (ą┤ą╗čÅ ą╝ąŠą┤ąĄą╗ąĖčĆąŠą▓ą░ąĮąĖčÅ ą┤ą░ą╗ąĄą║ąĖčģ ą▓ 菹▓ąŠą╗čÄčåąĖąŠąĮąĮąŠą╝ ą┐ą╗ą░ąĮąĄ ą▒ąĄą╗ą║ąŠą▓)
- 8. ą¤čĆąĄą┤čüą║ą░ąĘą░ąĮąĖąĄ ą▓č鹊čĆąĖčćąĮąŠą╣ čüčéčĆčāą║čéčāčĆčŗ http://www.new-science-press.com/ Table II of Williams, R.W. et al.: Biochimica et Biophysica Acta 1987, 916:200-204.
- 9. ŌĆō ą┐čĆąĄą┤čüą║ą░ąĘą░ąĮąĖąĄ ą┤ą╗čÅ ą║ą░ąČą┤ąŠą╣ ą░ą╝ąĖąĮąŠą║ąĖčüą╗ąŠčéčŗ ą▓ ą▓čŗą▒čĆą░ąĮąĮąŠą╝ ąŠą║ąĮąĄ čüąŠčüąĄą┤ąĮąĖčģ ą░ą╝ąĖąĮąŠą║ąĖčüą╗ąŠčé (13-21) ŌĆō čüą║ąŠčĆąĖąĮą│, ąŠą▒čāč湥ąĮąĖąĄ ą╝ąŠą┤ąĄą╗ąĖ ąĖ ą┐čĆąĄą┤čüą║ą░ąĘą░ąĮąĖąĄ 2D čüčéčĆčāą║čéčāčĆčŗ (ą╝ą░ą┐ą┐ąĖčĆąŠą▓ą░ąĮąĖąĄ 菹╗ąĄą╝ąĄąĮčéą░ ą▓č鹊čĆąĖčćąĮąŠą╣ čüčéčĆčāą║čéčāčĆčŗ ąĮą░ ąŠą║ąĮąŠ) ą¤čĆąĄą┤čüą║ą░ąĘą░ąĮąĖąĄ ą▓č鹊čĆąĖčćąĮąŠą╣ čüčéčĆčāą║čéčāčĆčŗ
- 10. ą£ąĄč鹊ą┤čŗ I. Chou-Fasman / GOR ą╝ąĄč鹊ą┤ II. ą£ąŠą┤ąĄą╗ąĖ ąĮąĄą╣čĆąŠąĮąĮčŗčģ čüąĄč鹥ą╣ III. ą£ąĄč鹊ą┤čŗ ┬½ą▒ą╗ąĖąČą░ą╣čłąĄą│ąŠ čüąŠčüąĄą┤ą░┬╗
- 11. ą£ąĄč鹊ą┤ Chou-Fasman (1974) ŌĆó ąĀą░ąĘčĆą░ą▒ąŠčéą░ąĮ Chou & Fasman ą▓ 1974 -1978 ŌĆó ąæą░ąĘą░ ŌĆō ąĖąĘą▓ąĄčüčéąĮčŗąĄ 3D čüčéčĆčāą║čéčāčĆčŗ ą│ą╗ąŠą▒čāą╗čÅčĆąĮčŗčģ ą▒ąĄą╗ą║ąŠą▓ ŌĆō ą¦ą░čüč鹊čéčŗ ą░ą╝ąĖąĮąŠą║ąĖčüą╗ąŠčé ą▓ ╬▒-čüą┐ąĖčĆą░ą╗čÅčģ ŌĆō ą¦ą░čüč鹊čéčŗ ą░ą╝ąĖąĮąŠą║ąĖčüą╗ąŠčé ą▓ ╬▓-ą╗ąĖčüčéą░čģ ŌĆō ą¦ą░čüč鹊čéčŗ ą░ą╝ąĖąĮąŠą║ąĖčüą╗ąŠčé ą▓ ╬▓-ą┐ąŠą▓ąŠčĆąŠčéą░čģ ŌĆō ą¤čĆą░ą▓ąĖą╗ą░ ąŠą▒čĆą░ąĘąŠą▓ą░ąĮąĖčÅ ╬▒-čüą┐ąĖčĆą░ą╗ąĄą╣ ąĖ ╬▓-ą╗ąĖčüč鹊ą▓ ŌĆó ą×čüąĮąŠą▓ą░ąĮ ąĮą░ čĆą░čüčéą▓ąŠčĆąĖą╝čŗčģ, ą│ą╗ąŠą▒čāą╗čÅčĆąĮčŗčģ ą▒ąĄą╗ą║ą░čģ ŌĆō ąĮą░čćą░ą╗čīąĮą░čÅ ą▒ą░ąĘą░ 15 ą▒ąĄą╗ą║ąŠą▓
- 13. ąĀą░ąĘą▓ąĖčéąĖąĄ Chou-Fasman 1. ą¤čĆąĖčüą▓ąŠąĄąĮąĖąĄ ą║ą░ąČą┤ąŠą╣ ą░ą╝ąĖąĮąŠą║ąĖčüą╗ąŠč鹥 ąŠą┐čĆąĄą┤ąĄą╗ąĄąĮąĮąŠą│ąŠ ą┐čāą╗ą░ ą┐ą░čĆą░ą╝ąĄčéčĆąŠą▓ 2. ąśą┤ąĄąĮčéąĖčäąĖą║ą░čåąĖčÅ a-helix ąĖ b-sheet. ąŻą┤ą╗ąĖąĮąĄąĮąĖąĄ čŹčéąĖčģ ąŠą▒ą╗ą░čüč鹥ą╣ ą▓ ąŠą▒ąŠąĖčģ ąĮą░ą┐čĆą░ą▓ą╗ąĄąĮąĖčÅčģ. 3. ą¤čĆąĖ ą┐ąĄčĆąĄą║čĆčŗčéąĖąĖ ŌĆō čüčĆą░ą▓ąĮąĄąĮąĖąĄ P(H) ąĖ P(E) ąĖ čüą║ąŠčĆąĖąĮą│.
- 14. 1. ąÆąĄčĆąŠčÅčéąĮąŠčüčéąĖ P╬▒(H)=[(#H in helix)/(#H)]/(fraction helix {all}) T S P T A E L M R S T G P(H) 69 77 57 69 142 151 121 145 98 77 69 57 P(E) 147 75 55 147 83 37 130 105 93 75 147 75 P(turn) 114 143 152 114 66 74 59 60 95 143 114 156 ąĀą░ąĘą▓ąĖčéąĖąĄ Chou-Fasman
- 15. ą¤ąŠąĖčüą║ a-čüą┐ąĖčĆą░ą╗ąĖ 2. ą¤ąŠąĖčüą║ ąŠą▒ą╗ą░čüč鹥ą╣, ą│ą┤ąĄ 4 ąĖąĘ 6 ą░ą╝ąĖąĮąŠą║ąĖčüą╗ąŠčé ąĖą╝ąĄčÄčé P(H) >100 (ŌĆ£ čÅą┤čĆąŠ a- čüą┐ąĖčĆą░ą╗ąĖŌĆØ) T S P T A E L M R S T G P(H) 69 77 57 69 142 151 121 145 98 77 69 57 T S P T A E L M R S T G P(H) 69 77 57 69 142 151 121 145 98 77 69 57
- 16. ąŻą┤ą╗ąĖąĮąĄąĮąĖąĄ čÅą┤čĆą░ a-čüą┐ąĖčĆą░ą╗ąĖ 3. ąĀą░čüčłąĖčĆąĄąĮąĖąĄ ąŠą▒ą╗ą░čüčéąĖ čÅą┤čĆą░, ą┐ąŠą║ą░ 4 ą░ą╝ąĖąĮąŠą║ąĖčüą╗ąŠčéčŗ ąĖą╝ąĄčÄčé čüčĆąĄą┤ąĮąĄąĄ P(H) >100. T S P T A E L M R S T G P(H) 69 77 57 69 142 151 121 145 98 77 69 57
- 17. ą¤ąŠąĖčüą║ ╬▓-ą╗ąĖčüčéą░ 4. ą¤ąŠąĖčüą║ ąŠą▒ą╗ą░čüč鹥ą╣, ą│ą┤ąĄ 3 ąĖąĘ 5 ą░ą╝ąĖąĮąŠą║ąĖčüą╗ąŠčé ąĖą╝ąĄčÄčé P(E) >100 ŌĆ£čÅą┤čĆąŠ ╬▓-ą╗ąĖčüčéą░ŌĆØ 5. ąŻą┤ą╗ąĖąĮąĄąĮąĖąĄ čÅą┤čĆą░ ą┤ąŠ č鹥čģ ą┐ąŠčĆ, ą┐ąŠą║ą░ 4 čüąŠčüąĄą┤ąĮąĖčģ ą░ą╝ąĖąĮąŠą║ąĖčüą╗ąŠčéčŗ ąĖą╝ąĄčÄčé čüčĆąĄą┤ąĮąĄąĄ P(E) > 100 6. ąĢčüą╗ąĖ score ąŠą▒ą╗ą░čüčéąĖ > 105 ąĖ čüčĆąĄą┤ąĮąĄąĄ P(E) > čüčĆąĄą┤ąĮąĄąĄ P(H), ąĘąĮą░čćąĖčé čŹčéą░ ąŠą▒ą╗ą░čüčéčī - ╬▓-ą╗ąĖčüčé T S P T A E L M R S T G P(H) 69 77 57 69 142 151 121 145 98 77 69 57 P(E) 147 75 55 147 83 37 130 105 93 75 147 75
- 18. GCG Programs ŌĆó PepPlot ŌĆō Plot on parallel panels ŌĆō -cff option, text output ŌĆó PeptideStructure ŌĆō text output (Most useful for detail) ŌĆó PlotStructure ŌĆō two outputs ŌĆó squiggles ŌĆ£protein-likeŌĆØ ŌĆó parallel panels
- 19. GOR III (Garnier-Osguthorpe-Robson. Gibrat J.F., J.Mol.Biol, 1987)
- 20. ą£ąŠą┤ąĄą╗ąĖ ąĮąĄą╣čĆąŠąĮąĮčŗčģ čüąĄč鹥ą╣ - ą£ą░čłąĖąĮąĮąŠąĄ ąŠą▒čāč湥ąĮąĖąĄ - ąĪąĄčé čüčéčĆčāą║čéčāčĆ (e.g. a-čüą┐ąĖčĆą░ą╗ąĖ, ąĮąĄ a-čüą┐ąĖčĆą░ą╗ąĖ) - ą×ą▒čāč湥ąĮąĖąĄ čĆą░čüą┐ąŠąĘąĮą░ą▓ą░čéčī čłą░ą▒ą╗ąŠąĮčŗ, čüčéčĆčāą║čéčāčĆčŗ ą▓ ąĖąĘą▓ąĄčüčéąĮčŗčģ ą▒ąĄą╗ą║ą░čģ ąŁčäč乥ą║čéąĖą▓ąĮąŠčüčéčī ~ 70 ŌĆō75% Rost B. Sander C. Prediction of Protein Secondary Structure at Better then 70% Accuracy. J.Mol.Biol., 1993, vol. 232. 584-599.NPS@ čüąĄčĆą▓ąĄčĆ
- 22. Eva
- 24. ą¤čĆąĄą┤čüą║ą░ąĘą░ąĮąĖąĄ ą▓č鹊čĆąĖčćąĮąŠą╣ čüčéčĆčāą║čéčāčĆčŗ Predict Protein (Mega) - secondary structure ( PHDsec, and PROFsec) PSI-pred (PSI-BLAST profiles used for prediction; David Jones, Warwick) PHD - Rost & Sander, EMBL, Germany ASPSSP server Raghava, INDIA DSC - King & Sternberg (this server) PREDATOR - Frischman & Argos (EMBL) ZPRED server Zvelebil et al., Ludwig, U.K. nnPredict Cohen et al., UCSF, USA. BMERC PSA Server Boston University, USA SSP (Nearest-neighbor) Solovyev and Salamov, Baylor College, USA. ŌĆó JPRED Consensus prediction (Cuff & Barton, EBI) ŌĆó NPS@
- 27. ą¤čĆąĄą┤čüą║ą░ąĘą░ąĮąĖąĄ čäčāąĮą║čåąĖąĖ ąĢčēąĄ ąŠą┤ąĮą░ ą▓ą░ąČąĮą░čÅ ąĘą░ą┤ą░čćą░ ą┐čĆąŠč鹥ąŠą╝ąĖą║ąĖ ŌĆö ą░ąĮą░ą╗ąĖąĘ ąĖ ą┐čĆąĄą┤čüą║ą░ąĘą░ąĮąĖąĄ čäčāąĮą║čåąĖąĖ ą▒ąĄą╗ą║ą░. ąśąĘą▓ąĄčüčéąĮąŠ, čćč鹊 čäčāąĮą║čåąĖčÅ ą▒ąĄą╗ą║ą░ ąŠą┐čĆąĄą┤ąĄą╗čÅąĄčéčüčÅ ąĄą│ąŠ ą░ą║čéąĖą▓ąĮčŗą╝ąĖ čüą░ą╣čéą░ą╝ąĖ, ą┐ąŠčŹč鹊ą╝čā ąĮą░ą║ąŠą┐ą╗ąĄąĮąĖąĄ ąĖ čüąĖčüč鹥ą╝ą░čéąĖąĘą░čåąĖčÅ ąĖąĮč乊čĆą╝ą░čåąĖąĖ ąŠą▒ ą░ą║čéąĖą▓ąĮčŗčģ čüą░ą╣čéą░čģ ą▒ąĄą╗ą║ąŠą▓ čćčĆąĄąĘą▓čŗčćą░ą╣ąĮąŠ ą░ą║čéčāą░ą╗čīąĮą░. ąÆ. ąśą▓ą░ąĮąĖčüąĄąĮą║ąŠ, ąö. ąōčĆąĖą│ąŠčĆąŠą▓ąĖč湥ą╝ ąĖ ąĪ. ą¤ąĖąĮčéčāčüąŠą╝ čĆą░ąĘčĆą░ą▒ąŠčéą░ąĮą░ ą║ąŠą╝ą┐čīčÄč鹥čĆąĮą░čÅ ą▒ą░ąĘą░ ą┤ą░ąĮąĮčŗčģ PDBSite, ą║ąŠč鹊čĆą░čÅ čüąŠą┤ąĄčƹȹĖčé ąĖąĮč乊čĆą╝ą░čåąĖčÄ ąŠ ą▒ąŠą╗ąĄąĄ č湥ą╝ 12 čéčŗčüčÅčćą░čģ ą░ą║čéąĖą▓ąĮčŗčģ čüą░ą╣č鹊ą▓ ą▒ąĄą╗ą║ąŠą▓. ąśčüč鹊čćąĮąĖą║ąŠą╝ ąĖąĮč乊čĆą╝ą░čåąĖąĖ čüą╗čāąČą░čé čģąŠčĆąŠčłąŠ ą┤ąŠą║čāą╝ąĄąĮčéąĖčĆąŠą▓ą░ąĮąĮčŗąĄ ą┐čĆąŠčüčéčĆą░ąĮčüčéą▓ąĄąĮąĮčŗąĄ čüčéčĆčāą║čéčāčĆčŗ ą▒ąĄą╗ą║ąŠą▓.
- 28. ŌĆó ą┐ą░čĆąĮąŠąĄ ą▓čŗčĆą░ą▓ąĮąĖą▓ą░ąĮąĖąĄ; ŌĆó ą╝ąĮąŠąČąĄčüčéą▓ąĄąĮąĮąŠąĄ ą▓čŗčĆą░ą▓ąĮąĖą▓ą░ąĮąĖąĄ; ŌĆó ą┐ąŠąĖčüą║ ą│ąŠą╝ąŠą╗ąŠą│ąŠą▓, threading; ŌĆó čüčéčĆčāą║čéčāčĆąĮąŠąĄ ą▓čŗčĆą░ą▓ąĮąĖą▓ą░ąĮąĖąĄ. ą×čüąĮąŠą▓ąĮčŗąĄ ą╝ąĄč鹊ą┤čŗ ą▓ ą▒ąĖąŠąĖąĮč乊čĆą╝ą░čéąĖą║ąĄ:
- 29. CASP Critical Assessment of Techniques for Protein Structure Prediction CASP1 (1994) CASP2 CASP3 CASP4 CASP5ŌĆ”..CASP9 (2010) ŌĆó Comparative modeling (CM) ŌĆó Fold-recognition (FR) ŌĆó CAFASP meta-server ver. 3 ŌĆó New folds (NF) ŌĆó Ten most wanted sec. struct. contacts, protein-protein docking, and disordered predictions.
- 30. About CASP: CASP is a blind study/experiment that aims at establishing the current state of the art in protein structure prediction; identifying what progress has been made; and highlighting where future effort may be most productively focused (Every two years). This blind study is held over an ~8 month time period and ends in a meeting held every two years, in Asilomar, CA, starting from 1994. For the procedure of the experiment, CASP participants are first provided target sequences (around May) via the Protein Structure Prediction Center at Lawrence Livermore National Laboratory. The participants have a few months to determine the template structure, alignment, model structure and evaluate their results. The sequence targets are categorized by homology and difficulty for predicting their structure. The fairly simple targets have med. sequence homology (>30% seq. identity) are considered comparative modeling (CM) predictions; the med. difficulty targets have med.-to-low sequence homology (~10- 30% seq. identity) are considered fold-recognition (FR) predictions; and the difficult targets have low seq. homology and usually require an ab initio methods are considered new folds (NF). During the prediction time (~May-Oct.), researchers (structural biologist in x-ray or NMR) work on solving the experimental structure of each of the target sequences and they hold back the structure coordinate information from the predictors. By Nov., all participants submit their models (as coordinates) to the Livermore Center and the researchers (who solve the target structure) finalize and post their results. Finally, in Dec., all participants and the CASP organizers meet to evaluate the results of the experiment comparing each model with the experimental structure and discussing the methodologies used. The goal of CAFASP is to evaluate the performance of fully automatic structure prediction servers available to the community. In contrast to the normal CASP procedure, CAFASP aims to answer the question of how well servers do without any intervention of experts, i.e. how well ANY user using only automated methods can predict protein structure. CAFASP assesses the performance of methods without the user intervention allowed in CASP.
- 32. ą¤čĆąĄą┤čüą║ą░ąĘą░ąĮąĖąĄ čüą▓ąŠčĆą░čćąĖą▓ą░ąĮąĖčÅ ą▒ąĄą╗ą║ą░ vs ą┐čĆąĄą┤čüą║ą░ąĘą░ąĮąĖąĄ čüčéčĆčāą║čéčāčĆčŗ ą¤čĆąĄčüčéą║ą░ąĘą░ąĮąĖąĄ ą┐čĆąŠčåąĄčüčüą░ č乊ą╗ą┤ąĖąĮą│ą░ ą▒ąĄą╗ą║ą░ čüą▓čÅąĘą░ąĮąŠ čü ą┐čĆąŠčåąĄčüčüąŠą╝ ą┐čĆąĖąŠą▒čĆąĄč鹥ąĮąĖčÅ ą▒ąĄą╗ą║ąŠą╝ ąĄą│ąŠ 3D č乊čĆą╝čŗ, ąŠč湥čĆčéą░ąĮąĖą╣ ŌĆō čäąĖąĘąĖą║ąŠ-čģąĖą╝ąĖč湥čüą║ąĖąĄ ą┐čĆąĖąĮčåąĖą┐čŗ. ą¤čĆąĄą┤čüą║ą░ąĘą░ąĮąĖąĄ čüčéčĆčāą║čéčāčĆčŗ ŌĆō ąĖčüą┐ąŠą╗čīąĘčāčÄčéčüčÅ ą╗čÄą▒čŗąĄ čüčéą░čéąĖčüčéąĖč湥čüą║ąĖąĄ, č鹥ąŠčĆąĄčéąĖč湥čüą║ąĖąĄ ąĖ 菹╝ą┐ąĖčĆąĖč湥čüą║ąĖąĄ ą┤ą░ąĮąĮčŗąĄ. 4 ą┐ąŠą┤čģąŠą┤ą░: ą£ąŠą┤ąĄą╗ąĖčĆąŠą▓ą░ąĮąĖąĄ ą│ąŠą╝ąŠą╗ąŠą│ąŠą▓ (Homology Modeling) ąĀą░čüą┐ąŠąĘąĮą░ą▓ą░ąĮąĖąĄ č乊ą╗ą┤ą░ (Sequence-Structure Threading (secondary structure prediction)): ŌĆó Dynamic programming ŌĆó Knowledge-based potentials ą¤čĆąĄą┤čüą║ą░ąĘą░ąĮąĖąĄ Ab initio Docking and Drug Design
- 33. ŌĆó ą£ąŠą┤ąĄą╗ąĖčĆąŠą▓ą░ąĮąĖąĄ ą│ąŠą╝ąŠą╗ąŠą│ąŠą▓ (homology modeling) ŌĆó Ab initio ą┐čĆąĄą┤čüą║ą░ąĘą░ąĮąĖąĄ ŌĆó ąĀą░čüą┐ąŠąĘąĮą░ą▓ą░ąĮąĖąĄ čüą▓ąŠčĆą░čćąĖą▓ą░ąĮąĖčÅ ŌĆ£Threading'ŌĆś ŌĆó ąöąŠą║ąĖąĮą│ ąóąĄčģąĮąĖą║ąĖ č乊ą╗ą┤ąĖąĮą│ą░ ą▒ąĄą╗ą║ą░
- 34. ąĪčĆą░ą▓ąĮąĖč鹥ą╗čīąĮąŠąĄ ą╝ąŠą┤ąĄą╗ąĖčĆąŠą▓ą░ąĮąĖąĄ ą│ąŠą╝ąŠą╗ąŠą│ąŠą▓ ąöą╗čÅ ą┐ąŠčüą╗ąĄą┤ąŠą▓ą░č鹥ą╗čīąĮąŠčüč鹥ą╣ čü ą│ąŠą╝ąŠą╗ąŠą│ąĖčćąĮąŠčüčéčīčÄ > 25-30% ąĖčüą┐ąŠą╗čīąĘąŠą▓ą░čéčī ąĖąĘą▓ąĄčüčéąĮčāčÄ PDB čüčéčĆčāą║čéčāčĆčā ą║ą░ą║ ąŠčéą┐čĆą░ą▓ąĮąŠą╣ ą┐čāąĮą║čé ą┤ą╗čÅ čüąŠąĘą┤ą░ąĮąĖčÅ 3D ą╝ąŠą┤ąĄą╗ąĖ čüčéčĆčāą║čéčāčĆčŗ ąĮąĄąĖąĘą▓ąĄčüčéąĮąŠą╣ ą┐ąŠčüą╗ąĄą┤ąŠą▓ą░č鹥ą╗čīąĮąŠčüčéąĖ. ąØčāąČąĮąŠ ąĖčüą┐ąŠą╗čīąĘąŠą▓ą░čéčī ą║ąŠąŠčĆą┤ąĖąĮą░čéčŗ ąŠčüąĮąŠą▓ąĮąŠą╣ čåąĄą┐ąĖ (N-C╬▒-C) ą│ąŠą╝ąŠą╗ąŠą│ąĖčćąĮąŠą╣ čüčéčĆčāą║čéčāčĆčŗ ą║ą░ą║ čłą░ą▒ą╗ąŠąĮ ą┤ą╗čÅ ą╝ąŠą┤ąĄą╗ąĖ 70% ąĖ ą▒ąŠą╗ąĄąĄ ą│ąŠą╝ąŠą╗ąŠą│ąĖčćąĮąŠčüčéąĖ ŌĆō ąŠč湥ąĮčī ą▓čŗčüąŠą║ąŠąĄ ą║ą░č湥čüčéą▓ąŠ ą╝ąŠą┤ąĄą╗ąĖ, ą┤ą░ąČąĄ ą┐ąŠą╗ąŠąČąĄąĮąĖčÅ ą▒ąŠą║ąŠą▓čŗčģ čåąĄą┐ąĄą╣ ą╝ąŠą│čāčé ą▒čŗčéčī ą┐čĆąĄą┤čüą║ą░ąĘą░ąĮčŗ čü ą▓čŗčüąŠą║ąŠą╣ č鹊čćąĮąŠčüčéčīčÄ. 40%-65% - čüčĆąĄą┤ąĮčÅčÅ č鹊čćąĮąŠčüčéčī ą┐čĆąĄą┤čüą║ą░ąĘą░ąĮąĖčÅ. ą£ąŠą│čāčé ą▒čŗčéčī čüąĄčĆčīčæąĘąĮčŗąĄ ąŠčłąĖą▒ą║ąĖ ą┤ą░ąČąĄ ą▓ ą┐ąŠą╗ąŠąČąĄąĮąĖąĖ ąŠčüąĮąŠą▓ąĮąŠą╣ čåąĄą┐ąĖ, ąŠčüąŠą▒ąĄąĮąĮąŠ ą▓ ąŠą▒ą╗ą░čüčéčÅčģ ą┐ąĄč鹥ą╗čīąĖąĘą│ąĖą▒ąŠą▓.
- 35. ąøąĄą║ą░čĆčüčéą▓ąĄąĮąĮčŗąĄ čüčĆąĄą┤čüčéą▓ą░, čĆą░ąĘčĆą░ą▒ąŠčéą░ąĮąĮčŗąĄ čü ąĖčüą┐ąŠą╗čīąĘąŠą▓ą░ąĮąĖąĄą╝ ąĮąĄą▓ąĄčĆąĮčŗčģ ą┐čĆąĄą┤čüčéą░ą▓ą╗ąĄąĮąĖą╣ ąŠ čüčéčĆčāą║čéčāčĆąĄ ą▒ąĄą╗ą║ą░, ą╝ąŠą│čāčé ą▒čŗčéčī č鹊ą║čüąĖčćąĮčŗ ąĖą╗ąĖ ąŠą▒ą╗ą░ą┤ą░čéčī ąĮąĄčāčćčéčæąĮąĮčŗą╝ąĖ ą┐ąŠą▒ąŠčćąĮčŗą╝ąĖ čŹčäč乥ą║čéą░ą╝ąĖ. ąöą╗čÅ čŹčäč乥ą║čéąĖą▓ąĮąŠčüčéąĖ čŹč鹊ą│ąŠ ą╝ąĄč鹊ą┤ą░ čéčĆąĄą▒čāąĄčéčüčÅ ą┐ąŠ ą╝ąĄąĮčīčłąĄą╣ ą╝ąĄčĆąĄ 3,000 čāąĮąĖą║ą░ą╗čīąĮčŗčģ, čüąŠą▓ąĄčĆčłąĄąĮąĮąŠ č鹊čćąĮąŠ ąŠą┐čĆąĄą┤ąĄą╗čæąĮąĮčŗčģ čüčéčĆčāą║čéčāčĆ. ąØą░ ą║ąŠąĮąĄčå 2001 ą│ąŠą┤ą░ ąĖą╝ąĄą╗ąŠčüčī č鹊ą╗čīą║ąŠ 1,000 čāąĮąĖą║ą░ą╗čīąĮčŗčģ čüčéčĆčāą║čéčāčĆ čüčĆąĄą┤ąĖ 16973 ą▓ PDB. 2008 ą│ąŠą┤ ŌĆō 53000 čüčéčĆčāą║čéčāčĆ ą▓ PDB, homo sapience ~1500.
- 36. 1. ą¤ąŠčüą╗ąĄą┤ąŠą▓ą░č鹥ą╗čīąĮąŠčüčéčī-čåąĄą╗čī ŌĆō ą┐ąĄčĆą▓ąĖčćąĮą░čÅ čüčéčĆčāą║čéčāčĆą░ ą▒ąĄą╗ą║ą░, 3D čüčéčĆčāą║čéčāčĆčā ą║ąŠč鹊čĆąŠą│ąŠ čüą╗ąĄą┤čāąĄčé ąŠą┐čĆąĄą┤ąĄą╗ąĖčéčī 2. ą©ą░ą▒ą╗ąŠąĮ ŌĆō ą▒ąĄą╗ąŠą║, čćčīčÅ 3D čüčéčĆčāą║čéčāčĆą░ čÅčüąĮą░ 3. ąÆčŗčĆą░ą▓ąĮąĖą▓ą░ąĮąĖąĄ ą┐ąŠčüą╗ąĄą┤ąŠą▓ą░č鹥ą╗čīąĮąŠčüč鹥ą╣ 1 ąĖ 2 ąĪčĆą░ą▓ąĮąĖč鹥ą╗čīąĮąŠąĄ ą╝ąŠą┤ąĄą╗ąĖčĆąŠą▓ą░ąĮąĖąĄ ą│ąŠą╝ąŠą╗ąŠą│ąŠą▓ ą¢ąĄą╗ą░č鹥ą╗čīąĮąŠ čéą░ą║ąČąĄ ąĖą╝ąĄčéčī ąæąĖąŠčģąĖą╝ąĖč湥čüą║čāčÄ ąĖ čüčéčĆčāą║čéčāčĆąĮčāčÄ ąĖąĮč乊čĆą╝ą░čåąĖčÄ (ą╗ąĖč鹥čĆą░čéčāčĆą░) ąöąŠą┐ąŠą╗ąĮąĖč鹥ą╗čīąĮčŗąĄ ą┐ąŠčüą╗ąĄą┤ąŠą▓ą░č鹥ą╗čīąĮąŠčüčéąĖ ą│ąŠą╝ąŠą╗ąŠą│ąŠą▓ čü ąĖąĘą▓ąĄčüčéąĮąŠą╣ čüčéčĆčāą║čéčāčĆąŠą╣
- 37. ą£ąŠą┤ąĄą╗ąĖčĆąŠą▓ą░ąĮąĖąĄ ą│ąŠą╝ąŠą╗ąŠą│ąŠą▓ 1. Fragment-based modeling: ąÆčŗčĆą░ą▓ąĮąĖą▓ą░ąĮąĖąĄ čü čåąĄą╗čīčÄ ąĖą┤ąĄąĮčéąĖčäąĖą║ą░čåąĖąĖ čüčéčĆčāą║čéčāčĆąĮąŠ- ą┐ąŠčüč鹊čÅąĮąĮčŗčģ ąŠą▒ą╗ą░čüč鹥ą╣ (SCR): ą░) ąŠą▒ą╗ą░čüčéąĖ ą▒ąĄąĘ ą▓čüčéą░ą▓ąŠą║- ą┤ąĄą╗ąĄčåąĖą╣ ąĖ ą▓) ąŠą▒ą╗ą░čüčéąĖ čü č湥čéą║ąŠ ąŠą┐čĆąĄą┤ąĄą╗čÅąĄą╝ąŠą╣ ą▓č鹊čĆąĖčćąĮąŠą╣ čüčéčĆčāą║čéčāčĆąŠą╣. VR ŌĆō ąŠą▒ą╗ą░čüčéąĖ ą╝ąĄąČą┤čā SCR. Composer (Sybil), Homology (InsightII) 2. Restraint-based modeling: ą┐ąŠą╗čāč湥ąĮąĖąĄ score-čäčāąĮą║čåąĖąĖ ą┐čāčéčæą╝ ą║ąŠą╝ą▒ąĖąĮąĖčĆąŠą▓ą░ąĮąĖčÅ ┬½ąŠą│čĆą░ąĮąĖč湥ąĮąĖą╣┬╗ - čĆą░čüčüč鹊čÅąĮąĖą╣ ą╝ąĄąČą┤čā ąĪ╬▒, č鹊čĆčüąĖąŠąĮąĮčŗčģ čāą│ą╗ąŠą▓ ąĖ čé.ą┤. ą×čåąĄąĮą║ą░ čĆąĄąĘčāą╗čīčéą░č鹊ą▓ MD ą┤ą░ąĮąĮąŠą╣ score- čäčāąĮą║čåąĖąĄą╣. Modeller
- 38. Swiss-PDBViewer
- 39. ŌĆó ą£ąŠą┤ąĄą╗ąĖčĆąŠą▓ą░ąĮąĖąĄ ą│ąŠą╝ąŠą╗ąŠą│ąŠą▓ ŌĆó ą¤čĆąĄą┤čüą║ą░ąĘą░ąĮąĖąĄ Ab initio ŌĆó ąĀą░čüą┐ąŠąĘąĮą░ą▓ą░ąĮąĖąĄ čüą▓ąŠčĆą░čćąĖą▓ą░ąĮąĖčÅ ŌĆ£Threading'ŌĆś ŌĆó ąöąŠą║ąĖąĮą│
- 40. ą¤čĆąĄą┤čüą║ą░ąĘą░ąĮąĖąĄ Ab initio ą¤čĆąĖą╝ąĄąĮčÅąĄčéčüčÅ, ą║ąŠą│ą┤ą░ ąĮąĄąĖąĘą▓ąĄčüčéąĮčŗ ą│ąŠą╝ąŠą╗ąŠą│ąĖ, ąĮąĄčé čüčéčĆčāą║čéčāčĆčŗ, ą║ąŠč鹊čĆčāčÄ ą╝ąŠąČąĮąŠ ą▒čŗą╗ąŠ ą▒čŗ ąĖčüą┐ąŠą╗čīąĘąŠą▓ą░čéčī ą║ą░ą║ čłą░ą▒ą╗ąŠąĮ ąĢčüčéčī č鹊ą╗čīą║ąŠ ąŠą┤ąĮą░ ą┐ąŠčüą╗ąĄą┤ąŠą▓ą░č鹥ą╗čīąĮąŠčüčéčī. ą¤čĆąĄą┤čüą║ą░ąĘą░ąĮąĖąĄ 3D ąŠčüąĮąŠą▓ą░ąĮąŠ ąĮą░ ┬½ą▒ą░ąĘąŠą▓čŗčģ┬╗ ą┐čĆąĖąĮčåąĖą┐ą░čģ, čéą░ą║ąĖčģ, ą║ą░ą║ 菹ĮąĄčĆą│ąĄčéąĖč湥čüą║ąĖąĄ ąĖ čüčéą░čéąĖčüčéąĖč湥čüą║ąĖąĄ ąĘą░ą║ąŠąĮčŗ ąĖ ą┐čĆą░ą▓ąĖą╗ą░. ąŁč鹊 ŌĆō čüąĖą╝čāą╗čÅčåąĖčÅ čäąĖąĘąĖč湥čüą║ąĖčģ čüąĖą╗ ąĖ ą┐čĆąŠčåąĄčüčüąŠą▓, ą║ąŠč鹊čĆčŗąĄ ą╝ąŠą│čāčé ą┐čĆąĖą▓ąĄčüčéąĖ čĆą░ąĘą▓čæčĆąĮčāčéčŗą╣ ą▒ąĄą╗ąŠą║ ą▓ ąĮą░čéąĖą▓ąĮčāčÄ (čüčéą░ą▒ąĖą╗čīąĮčāčÄ, ą┐čĆąĖčüčāčéčüčéą▓čāčÄčēčāčÄ ą▓ ą┐čĆąĖčĆąŠą┤ąĄ) ą║ąŠąĮč乊čĆą╝ą░čåąĖčÄ ąĮą░ ą║ąŠą╝ą┐čīčÄč鹥čĆąĄ ąĪčéą░ą▒ąĖą╗čīąĮąŠčüčéčī čü č鹊čćą║ąĖ ąĘčĆąĄąĮąĖčÅ č鹥čĆą╝ąŠą┤ąĖąĮą░ą╝ąĖą║ąĖ: ąĮą░čéąĖą▓ąĮą░čÅ ą║ąŠąĮč乊čĆą╝ą░čåąĖčÅ ą▒ąĄą╗ą║ą░ ąĄčüčéčī ąĄą│ąŠ ą│ą╗ąŠą▒ą░ą╗čīąĮčŗą╣ ą╝ąĖąĮąĖą╝čāą╝ čüą▓ąŠą▒ąŠą┤ąĮąŠą╣ 菹ĮąĄčĆą│ąĖąĖ. ąæąĄą╗ąŠą║ ą┤ąŠą╗ąČąĄąĮ čüą▓ąŠčĆą░čćąĖą▓ą░čéčīčüčÅ čéą░ą║ čüą░ą╝ąŠčüč鹊čÅč鹥ą╗čīąĮąŠ.
- 41. ą¤čĆąĄą┤čüą║ą░ąĘą░ąĮąĖąĄ Ab initio ąŁą╗ąĄą║čéčĆąŠčüčéą░čéąĖč湥čüą║ąĖąĄ ąÆą░ąĮ-ąöąĄčĆ-ąÆą░ą░ą╗čīčü ąÆąŠą┤ąŠčĆąŠą┤ąĮčŗąĄ čüą▓čÅąĘąĖ ąŁąĮąĄčĆą│ąĖčÅ č鹊čĆčüąĖąŠąĮąĮčŗčģ čüą▓čÅąĘąĄą╣
- 42. ą¤čĆąĄą┤čüą║ą░ąĘą░ąĮąĖąĄ Ab initio - čüą▓ąŠčĆą░čćąĖą▓ą░ąĮąĖąĄ ą¤ąŠą╗ąĮčŗą╣ čĆą░čüčćčæčé 菹ĮąĄčĆą│ąĖą╣ ŌĆō ąŠč湥ąĮčī ąĘą░čéčĆą░čéąĮčŗą╣ čü č鹊čćą║ąĖ ąĘčĆąĄąĮąĖčÅ ą▓čŗčćąĖčüą╗ąĄąĮąĖą╣ ą┐čĆąŠčåąĄčüčü. ą¤ąŠčŹč鹊ą╝čā čéčĆąĄą▒čāąĄčéčüčÅ čĆą░ąĘčĆą░ą▒ąŠčéą║ą░ ąĮąĄą║ąĖčģ 菹▓čĆąĖčüčéąĖč湥čüą║ąĖčģ 菹ĮąĄčĆą│ąĄčéąĖč湥čüą║ąĖčģ čäčāąĮą║čåąĖą╣, ą║ąŠč鹊čĆčŗąĄ ą▒čŗ ąĮą░ą┤čæąČąĮąŠ čĆą░ąĘą╗ąĖčćą░ą╗ąĖ ┬½ą┐čĆą░ą▓ąĖą╗čīąĮčāčÄ┬╗ ąĖ ┬½ąĮąĄą┐čĆą░ą▓ąĖą╗čīąĮčāčÄ┬╗ čüčéčĆčāą║čéčāčĆčŗ ąĖ ą╗čāčćčłąĄ ┬½ą┐ąŠąĮąĖą╝ą░ą╗ąĖ┬╗ ą▒čŗ čüąĖą╗čŗ, ą║ąŠč鹊čĆčŗąĄ čāą┐čĆą░ą▓ą╗čÅčÄčé čüą▓ąŠčĆą░čćąĖą▓ą░ąĮąĖąĄą╝ ą▒ąĄą╗ą║ą░.
- 44. ążąŠą╗ą┤ąĖąĮą│. ą¤čĆąĄą┤čüą║ą░ąĘą░ąĮąĖąĄ Ab initio Protein Folding: A Perspective from Theory and Experiment Christopher M. Dobson,* Andrej S┼ō ali, and Martin Karplus*
- 45. ą¤čĆąĄą┤čüą║ą░ąĘą░ąĮąĖąĄ Ab initio ąĪčĆą░ą▓ąĮąĄąĮąĖąĄ čĆą░čüčćčæčéąĮąŠą╣ ąĖ 菹║čüą┐ąĄčĆąĖą╝ąĄąĮčéą░ą╗čīąĮąŠą╣ ą╝ąŠą┤ąĄą╗ąĖ ą┤ą╗čÅ ą▒ąĄą╗ą║ą░ ą╝ąĖąŠą│ą╗ąŠą▒ąĖąĮą░ ąĖ ąĖčüą┐ąŠą╗čīąĘąŠą▓ą░ąĮąĖąĄą╝ refined potential function. ąĀą░čüčüčćąĖčéą░ąĮąĮą░čÅ čüčéčĆčāą║čéčāčĆą░ čÅą▓ą╗čÅąĄčéčüčÅ 3D čüčéčĆčāą║čéčāčĆąŠą╣, ą┐ąŠą╗čāč湥ąĮąĮąŠą╣ ą▓ čĆąĄąĘčāą╗čīčéą░č鹥 3-čģ čĆą░ąĘąĮčŗčģ čĆą░čüčćčæč鹊ą▓ čü ą┤ą░ą╗čīąĮąĄą╣čłąĄą╣ ą║ą╗ą░čüč鹥čĆąĖąĘą░čåąĖąĄą╣ ąĖ ą▓čŗą▒ąŠčĆąŠą╝ čüčéčĆčāą║čéčāčĆčŗ čü ąĮą░ąĖą╝ąĄąĮčīčłąĄą╣ 菹ĮąĄčĆą│ąĖąĄą╣. ą×ą▒čēąĄąĄ ą▓čĆąĄą╝čÅ čüąĖą╝čāą╗čÅčåąĖąĖ ąĮą░ ą║ą╗ą░čüč鹥čĆąĄ ąĖąĘ 16 ą╝ą░čłąĖąĮ CM-5 massively parallel computer čüąŠčüčéą░ą▓ąĖą╗ąŠ 60 čćą░čüąŠą▓, ą▓ č鹥č湥ąĮąĖąĖ ą║ąŠč鹊čĆčŗčģ ą▒čŗą╗ąŠ ą│ąĄąĮąĄčĆąĖčĆąŠą▓ą░ąĮąŠ ą┐ąŠčĆčÅą┤ą║ą░ 5 ą╝ąĖą╗ą╗ąĖąŠąĮąŠą▓ čüčéčĆčāą║čéčāčĆ. RMS čüąŠčüčéą░ą▓ą╗čÅąĄčé 6.2 ├ģ.
- 46. ą¤ą░čĆą░ą┤ąŠą║čü ąøąĄą▓ąĄąĮčéą░ą╗čÅ ąÆčĆąĄą╝čÅ, ąĘą░ ą║ąŠč鹊čĆąŠąĄ ą▒ąĄą╗ąŠą║ čüą║čĆčāčćąĖą▓ą░ąĄčéčüčÅ, (ą┐čĆąĖąĮąĖą╝ą░ąĄčé ą║ąŠąĮąĄčćąĮąŠąĄ 3D čüąŠčüč鹊čÅąĮąĖąĄ) ąĮą░ ą╝ąĮąŠą│ąŠ ą┐ąŠčĆčÅą┤ą║ąŠą▓ ą╝ąĄąĮčīčłąĄ ą▓čĆąĄą╝ąĄąĮąĖ ą┐ąĄčĆąĄą▒ąŠčĆą░ ą▓čüąĄčģ ą▓ąŠąĘą╝ąŠąČąĮčŗčģ ą║ąŠąĮčäąĖą│čāčĆą░čåąĖą╣. ąöąŠą┐čāčüčéąĖą╝, ą▓ ą▒ąĄą╗ą║ąĄ 100 ą░č鹊ą╝ąŠą▓, ą║ą░ąČą┤čŗą╣ ąĖąĘ ą║ąŠč鹊čĆčŗčģ ą┐čĆąĖąĮąĖą╝ą░ąĄčé 3 ą┐ąŠą╗ąŠąČąĄąĮąĖčÅ: 3 100 = 5 ├Ś 10 47 ą║ąŠąĮč乊čĆą╝ą░čåąĖą╣. ąØą░ąĖą▒čŗčüčéčĆąĄą╣čłąĄąĄ ą┤ą▓ąĖąČąĄąĮąĖąĄ ŌĆō 10- 15 čü. ą¤ąĄčĆąĄą▒ąŠčĆ ą▓čüąĄčģ ą║ąŠąĮč乊čĆą╝ą░čåąĖą╣ ąĘą░ą╣ą╝čæčé 5 ├Ś 10 32 čü ąĖą╗ąĖ 1.6 ├Ś 10 25 ą╗ąĄčé (ą▓ąŠąĘčĆą░čüčé ąÆčüąĄą╗ąĄąĮąĮąŠą╣ ~ 13,75 ├Ś 109 )
- 47. ŌĆó Homology Modeling ŌĆó Ab initio prediction ŌĆó Fold Recognition or ŌĆ£Threading'ŌĆś
- 48. ąĀą░čüą┐ąŠąĘąĮą░ą▓ą░ąĮąĖąĄ čüą▓ąŠčĆą░čćąĖą▓ą░ąĮąĖčÅ (ŌĆ£ThreadingŌĆØ) ąØą░ą┐ąŠą╝ąĖąĮą░ąĄčé ą╝ąĄč鹊ą┤ ą╝ąŠą┤ąĄą╗ąĖčĆąŠą▓ą░ąĮąĖčÅ ą│ąŠą╝ąŠą╗ąŠą│ąŠą▓, ąĮąŠ ąĮąĄ čéčĆąĄą▒čāąĄčé čüčéčĆčāą║čéčāčĆ čü ą▓čŗčüąŠą║ąŠą╣ čüč鹥ą┐ąĄąĮčīčÄ ąĖą┤ąĄąĮčéąĖčćąĮąŠčüčéąĖ. ąśąĮč鹥čĆąĄčüčāčÄčēą░čÅ ąĮą░čü ą┐ąŠčüą╗ąĄą┤ąŠą▓ą░č鹥ą╗čīąĮąŠčüčéčī ┬½ą┐čĆąŠčéčÅą│ąĖą▓ą░ąĄčéčüčÅ┬╗ č湥čĆąĄąĘ ą▓čüąĄ ą▓ąŠąĘą╝ąŠąČąĮčŗąĄ ą┐ąŠąĘąĖčåąĖąĖ ąŠčüąĮąŠą▓ąĮąŠą╣ čåąĄą┐ąĖ ą▓ąŠ ą▓čüąĄčģ ąĖąĘą▓ąĄčüčéąĮčŗčģ ą▒ąĄą╗ą║ąŠą▓čŗčģ čüčéčĆčāą║čéčāčĆą░čģ ą▓ PDB, ąĖ ą┤ą╗čÅ ą║ą░ąČą┤ąŠą╣ ąĖč鹥čĆą░čåąĖąĖ čĆą░čüčüčćąĖčéčŗą▓ą░ąĄčéčüčÅ ąĄčæ čüą▓ąŠą▒ąŠą┤ąĮą░čÅ čŹąĮąĄčĆą│ąĖčÅ. ąĪčéčĆčāą║čéčāčĆą░, ą║ąŠč鹊čĆą░čÅ ą┤ą░čüčé ą╗čāčćčłąĖą╣ ą┐ąŠą║ą░ąĘą░č鹥ą╗čī 菹ĮąĄčĆą│ąĖąĖ ą┐čĆąĖąĮąĖą╝ą░ąĄčéčüčÅ ąĘą░ ┬½čłą░ą▒ą╗ąŠąĮ┬╗ ąĖ ą┤ą░ą╗čīąĮąĄą╣čłąĖą╣ ą┐čĆąŠčåąĄčüčü ąĮą░ą┐ąŠą╝ąĖąĮą░ąĄčé ą╝ąŠą┤ąĄą╗ąĖčĆąŠą▓ą░ąĮąĖąĄ ą│ąŠą╝ąŠą╗ąŠą│ąŠą▓ Threading ąĮąĄ ą╝ąŠąČąĄčé ą▒čŗčéčī ą┐čĆąĖą╝ąĄąĮčæąĮ ą┤ą╗čÅ č鹥čģ ą▒ąĄą╗ą║ąŠą▓, ą┤ą╗čÅ ą║ąŠč鹊čĆčŗčģ ą▓ ą▒ą░ąĘąĄ PDB ąĮąĄčé ą┐ąŠčģąŠąČąĖčģ čüčéčĆčāą║čéčāčĆ.
- 49. ąśąĘ ┬½Methods in Molecular Biology, vol 143, Methods and ProtocolMethods and Protocols. Protein Structure Prediction, ąĄdited by David M. Webster┬╗ Profiles-3D scoring function: ąŠčåąĄąĮą║ą░ ą╗ąŠą║ą░ą╗čīąĮąŠą│ąŠ čüčéčĆčāą║čéčāčĆąĮąŠą│ąŠ ą▓čŗčĆą░ą▓ąĮąĖą▓ą░ąĮąĖčÅ (čāą║ą╗ą░ą┤ą║ąĖ) ą║ą░ąČą┤ąŠą╣ ą░ą╝ąĖąĮąŠą║ąĖčüą╗ąŠčéčŗ ą▓ ą┐ąŠčüą╗ąĄą┤ąŠą▓ą░č鹥ą╗čīąĮąŠ- čüčéąĖ ą▒ąĄąĘ čāč湥čéą░ ą┐ąŠą┐ą░čĆąĮąŠą│ąŠ ą▓ąĘą░ąĖą╝ąŠą┤ąĄą╣- čüčéą▓ąĖčÅ ą░ą╝ąĖąĮąŠą║ąĖčüą╗ąŠčé+čüą║ą╗ąŠąĮąĮąŠčüčéčī ą║ H/E/L čüčéčĆčāą║čéčāčĆą░ą╝+ą┐ąŠą╗čÅčĆąĮąŠčüčéčī (solvent exposure) ąĀą░čüą┐ąŠąĘąĮą░ą▓ą░ąĮąĖąĄ čüą▓ąŠčĆą░čćąĖą▓ą░ąĮąĖčÅ (ŌĆ£ThreadingŌĆØ)
- 50. ąĀąĖčüčāąĮąŠą║ ąĖąĘ R. Lathrop et al, ŌĆ£Analysis and Algorithms for Protein Sequence-Structure AlignmentŌĆØ in Computational Methods in Molecular Biology, Salzberg et al. editors, 1998. ąĀą░čüą┐ąŠąĘąĮą░ą▓ą░ąĮąĖąĄ čüą▓ąŠčĆą░čćąĖą▓ą░ąĮąĖčÅ (ŌĆ£ThreadingŌĆØ)
- 51. Fold Recognition ŌĆō The Fold PDB Groups clustered by a common resemblanc e Genome Sequencing Homology Structure Conservation Calculated Folds ąĪą║ąŠą╗čīą║ąŠ ą▓čüąĄą│ąŠ č乊ą╗ą┤ąŠą▓? ąÜąŠą╗ąĖč湥čüčéą▓ąŠ č乊ą╗ą┤ąŠą▓ ~ 4000 ąæąö ąĖąĘ 930 č乊ą╗ą┤ąŠą▓ ~ 90% čüąĄą╝ąĄą╣čüčéą▓ ą▒ąĄą╗ą║ąŠą▓
- 52. Fold Recognition ŌĆō ąĮąĄą┤ąŠčüčéą░čéą║ąĖ ąŁč鹊čé ą╝ąĄč鹊ą┤ čĆąĄą┤ą║ąŠ ą┐čĆąĖą▓ąŠą┤ąĖčé ą║ č鹊ą╝čā ą║ą░č湥čüčéą▓čā čüčéčĆčāą║čéčāčĆąĮąŠą│ąŠ ą▓čŗčĆą░ą▓ąĮąĖą▓ą░ąĮąĖčÅ, ą║ąŠč鹊čĆąŠąĄ ą┐čĆąĄą┤ąŠčüčéą░ą▓ą╗čÅąĄčé ą╝ąŠą┤ąĄą╗ąĖčĆąŠą▓ą░ąĮąĖąĄ ą│ąŠą╝ąŠą╗ąŠą│ąŠą▓.
- 53. ąĪąĄčĆą▓ąĄčĆčŗ ŌĆóPredictProtein Server ŌĆóModBase (a database of three-dimensional protein models calculated by comparative modeling( 3D PSSM & ModBase 3D-PSSM ą┐čĆąĄą┤čüą║ą░ąĘą░ąĮąĖąĄ 3D čüčéčĆčāą║čéčāčĆčŗ ą┐ąŠ ą┐ąŠčüą╗ąĄą┤ąŠą▓ą░č鹥ą╗čīąĮąŠčüčéąĖ ąĖ ą▓ąĄčĆąŠčÅčéąĮąŠčüčéčī čŹč鹊ą╣ čüčéčĆčāą║čéčāčĆčŗ ModBase ŌĆō ą▒ą░ąĘą░ ą┤ą░ąĮąĮčŗčģ 3D čüčéčĆčāą║čéčāčĆ, ą┐ąŠčüčéčĆąŠąĄąĮąĮčŗčģ ąĮą░ ąŠčüąĮąŠą▓ąĄ čüčĆą░ą▓ąĮąĖč鹥ą╗čīąĮąŠą│ąŠ ą╝ąŠą┤ąĄą╗ąĖčĆąŠą▓ą░ąĮąĖčÅ
Editor's Notes
- #3: 1
- #5: Experimental data can aid the structure prediction process. Some of these are: Disulphide bonds, which provide tight restraints on the location of cysteines in space Spectroscopic data, which can give you and idea as to the secondary structure content of your protein Site directed mutagenesis studies, which can give insights as to residues involved in active or binding sites Knowledge of proteolytic cleavage sites, post-translational modifictions, such as phosphorylation or glycosylation can suggest residues that must be accessible Protein Sequence: Transmembrane? Coil-coil? Does your protein contain regions of low complexity? Proteins frequently contain runs of poly-glutamine or poly-serine, which do not predict well (SEG program). If the answer to any of the above questions is yes, then it is worthwhile trying to break your sequence into pieces, or ignore particular sections of the sequence, etc. This is related to the problem of locating domains .
- #7: Fig.:Coverage for each species is reported as the fraction of the residues in the proteome that are annotated . Structural annotation is an homology to a known structure. Functional annotation is when there is no structural annotation but there is an homology to a sequence database entry that has a useful description. Homology denotes a sequence similarity to a structurally or functionally un-annotated protein, such as one described as hypothetical. Non-globular denotes remaining sequence regions that were predicted as transmembrane, signal peptide, coiled-coils or low-complexity. Remaining residues are classified as orphans.
- #9: Analysis of the frequency with which different amino acids are found in different types of secondary structure shows some general preferences. For example, long side chains such as those of leucine, methionine, glutamine and glutatamic acid are often found in helices, presumably because these extended side chains can project out away from the crowded central region of the helical cylinder. In contrast, residues whose side chains are branched at the beta carbon , such as valine, isoleucine and phenylalanine are more often found in beta sheets, because every other side chain in a sheet is pointing in the opposite direction, leaving room for beta-branched side chains to pack. Such tendencies underlie various empirical rules for the prediction of secondary structure from sequence, such as those of Chou and Fasman. In the Chou-Fasman and other statistical methods of predicting secondary structure, the assumption is made that local effects predominate in determining whether a stretch of sequence will be helical, form a turn, compose a beta strand, or adopt an irregular conformation. This assumption is probably only partially valid, which may account for the failure of such methods to achieve close to 100% success in secondary structure prediction. The methods take proteins of known three-dimensional structure and tabulate the preferences of individual amino acids for various structural elements. By comparing these values with what might be expected randomly, conformational preferences can be assigned to each amino acid. To apply these preferences to a sequence of unknown structure, a moving window of about five residues is scanned along a sequence, and the average preferences are tallied. Empirical rules are then applied to assign secondary structural features based on the average preferences. Unfortunately, these tendencies are only very rough, and there are many exceptions. It is probably more useful to consider which side chains are disfavored in particular types of secondary structures. With specialized exceptions Proline is disfavored in both helices and sheets because it has no backbone N-H group to participate in hydrogen bonding. Glycine is also less commonly found in helices and sheets, in part because it lacks a side chain and therefore can adopt a much wider range of phi, psi torsion angles in peptides. These two residues are, however, strongly associated with beta turns, and sequences such as Pro-Gly and Gly-Pro are sometimes considered diagnostic for turns. Although predictive schemes based on residue preferences have some value, none is completely accurate, and the one rule that seems to be most reliable is that any amino acid can be found in any type of secondary structure, if only infrequently. Proline, for instance, is sometimes found in alpha helices; when it is, it simply interrupts the helical hydrogen-bonding network and produces a kink in the helix.
- #11: 1974 Chou and Fasman propose a statistical method based on the propensities of amino acids to adopt secondary structures based on the observation of their location in 15 protein structures determined by X-ray diffraction. Clearly these statistics derive from the particular stereochemical and physicochemical properties of the amino acids. See for example, glycine and proline. These statistics have been refined over the years by a number of authors (including Chou and Fasman themselves) using a larger set of proteins. Rather than a position by position analysis the propensity of a position is calculated using an average over 5 or 6 residues surrounding each position. On a larger set of 62 proteins the base method reports a success rate of 50%. 1978 Garnier improved the method by using statistically significant pair-wise interactions as a determinant of the statistical significance. This improved the success rate to 62% 1993 Levin improved the prediction level by using multiple sequence alignments. The reasoning is as follows. Conserved regions in a multiple sequence alignment provides a strong evolutionary indicator of a role in the function of the protein. Those regions are also likely to have conserved structure, including secondary structure and strengthen the prediction by their joint propensities. This improved the success rate to 69%. 1994 Rost and Sander combined neural networks with multiple sequence alignments. The idea of a neural net is to create a complex network of interconnected nodes, where progress from one node to the next depends on satisfying a weighted function that has been derived by training the net with data of known results, in this case protein sequences with known secondary structures. The success rate is 72%.
- #22: Simulate the brain. Selection of training sets is extremely important. Different protein families, only one or two representative from each family.
- #23: Jpred: (http://www.dl.ac.uk/CCP/CCP11/newsletter/vol2_4/jpred_ccp11/) Jpred runs DSC (5), PHD (1,2), PREDATOR (3,4) and NNSSP (6) in parallel to build its consensus prediction, but predictions from slightly less accurate algorithms MULPRED (8) and ZPRED (7) are also included in the final output.╠²╠²╠² These methods were chosen as representatives of current state-of-the-art secondary structure predictions methods that exploit the evolutionary information from multiple sequences.╠² Each derives its prediction using a different heuristic, based upon nearest neighbours (NNSSP), jury decision neural networks (PHD), linear discrimination (DSC), consensus single sequence method (MULPRED), hydrogen bonding propensities (PREDATOR), or conservation number weighted prediction (ZPRED).╠²╠²╠²╠² The consensus is constructed using a simple majority wins combination of DSC, PHD, PREDATOR and NNSSP, relying on the PHD prediction if there is a a tie.╠² In our study, we found this combination to be optimal.╠²
- #24: Flowchart of EVA. Every day, EVA downloads the newest protein structures from PDB [1] . The structures are added to mySQL databases, sequences are extracted for every protein chain, and are sent to each prediction server by META-PredictProtein [2] . META-PP collects the results and sends them to EVA. Every week, EVA runs alignment programs for searching sequence (iterated PSI-BLAST [3] , MaxHom [4] ) and structure (CE [5] , ProSub [6] ) databases to determine homologues. Predictions of secondary structure and inter-residue contacts, as well as, comparative modelling are evaluated at the EVA satellites at Columbia University, Rockefeller University, and CNB Madrid. Goals: CASP addresses the question ŌĆśhow well can experts predict protein structure if given sufficient incentive to do so?ŌĆÖ. In contrast, the question addressed by EVA is ŌĆśhow well could molecular biologists predict protein structure, if they simply take the output from the programs out there?ŌĆÖ. Thus, the goals are: Provide a continuous, fully automated, and statistically significant analysis of structure prediction servers. As has been shown by many of us, predictions based on small numbers of samples are NOT representative. EVA running for a year could produce a fairly representative picture. Even running for a month EVA could produce more reliable estimates than CASP can do in 2 years (at least, for answering the particular, restricted ŌĆö but important - question ŌĆśhow well do servers doŌĆÖ). EVA will NOT answer to requests of users!! It will NOT be a meta-server, rather it will simply sit there and evaluate servers based on known structures. EVA will NOT evaluate any server without the consent of the author.
- #26: SSE ŌĆō secondary structure elements Perutz (1990) showed (while working with hemoglobin and myoglobin) that amphipathicity can be detected in the sequnce: non polar residues can appears every 3.6 approx. in a linear sequence, making one side of the helix hydrophobic.
- #27: An example of the prediction of secondary structure from sequence for a protein of unknown function from the Enterococcus faecalis genome. What is striking is that all of the schemes agree on the approximate locations of the alpha helices (h) and beta strands (e), but they disagree considerably on the lengths and end positions of these segments. Note also that the probable positions of loops (indicated by a c) and turns (indicated by a t) are very inconsistently predicted. Such results are typical, but the application of many methods is clearly more informative than the use of a single one. The bottom line shows the consensus prediction.
- #28: The upper one is by Jpred and the lower one is by GOR
- #40: 1wqa, 1tx4, 1grn, 1tad and 1gfi. -> only 1tx4, 1grn, 1tad Note that some amino acids may appear in yellow once a molecule has been loaded. It signifies that their sidechain has been reconstructed during the loading process because some atoms were lacking. When all sidechain atoms are lacking, a rotamer library is searched until the rotamer that generate a maximum of H-bonds and a minimum of steric hindrances is found. If only some sidechain atoms are lacking, the rotamer that gives the lowest RMS when fitted to the partial sidechain is taken. In any case, you may try to find a better sidechain manually with the mutation tool. If you want to act on a complete column , simply hold down the shift key while clicking in a column Note: if a little earth icon is shown below the first tool, the rotation takes place in absolute coordinates. Otherwise (little protein icon) molecules are rotated around their centrotid. Hence the first option allows you to rotate the molecule around any atom, providing that this atom has previously been centered (translated to the (0,0,0) coordinate). Note: if "caps lock" is down, you can measure several distances or angles successively. To exit the "repeated" measurement mode, you can either depress "caps lock" or hit "esc".
- #53: ąŻčüčéąĮčŗąĄ ą┐ąŠčÅčüąĮąĄąĮąĖčÅ.