


































More Related Content
Similar to Density Functional Theory by Tayyab Shabir (20)
More from TayyabShabir (6)




Recently uploaded (20)




















Density Functional Theory by Tayyab Shabir
- 1. 1 Density Functional Theory (DFT) ’üČ 1998: Nobel prize awarded to Walter Kohn Walter Kohn a Austrian- American theoretical physicist and theoretical chemist ’üČ To solve many body problems by Schr├Čdinger's equation. H’ü╣ = E’ü╣
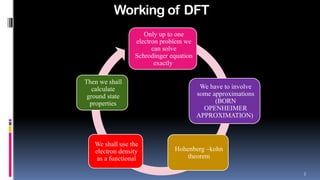
- 2. 2 Working of DFT Only up to one electron problem we can solve Schrodinger equation exactly We have to involve some approximations (BORN OPENHEIMER APPROXIMATION) Hohenberg ŌĆōkohn theorem We shall use the electron density as a functional Then we shall calculate ground state properties
- 3. ’é¦ The Hamiltonian for N-Particle system ’é¦ BORN OPPENHEIMER APPROXIMATION ØæÜØæøØæóØæÉØæÖØæÆØæ¢ Ōē½ØæÜØæÆ ŌĆó Reduced dimension from 3Ne to 3 by considering nuclei is static. ’é¦ HOHENBERG ŌĆō KOHN THEOREMS Theorem: 1 ŌĆ£The external potential vext or the ground state energy E is a unique functional of electron densityŌĆØ. Theorem: 2 ŌĆ£The electron density that minimizes the energy of the overall functional is the true ground state electron densityŌĆØ. Limitations of HK Theorems They do provide method of finding in practice however these theorems were not very helpful in real calculation. Two other scientists Kohn and Sham gave an equation which turned DFT into a practical tool. 17
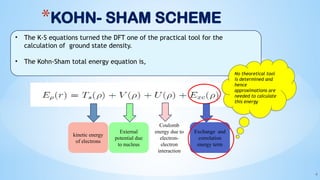
- 4. *KOHN- SHAM SCHEME 4 ŌĆó The K-S equations turned the DFT one of the practical tool for the calculation of ground state density. ŌĆó The Kohn-Sham total energy equation is, kinetic energy of electrons External potential due to nucleus Coulomb energy due to electron- electron interaction Exchange and correlation energy term No theoretical tool is determined and hence approximations are needed to calculate this energy
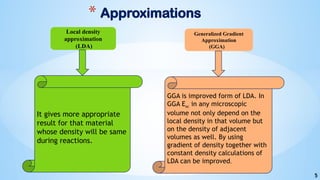
- 5. 5 * Approximations Local density approximation (LDA) Generalized Gradient Approximation (GGA) It gives more appropriate result for that material whose density will be same during reactions. GGA is improved form of LDA. In GGA Exc in any microscopic volume not only depend on the local density in that volume but on the density of adjacent volumes as well. By using gradient of density together with constant density calculations of LDA can be improved.
- 6. 6 *Amsterdam Density Functional ŌĆó Developed in 1970 ŌĆó Vrije University of Amsterdam and university of Calgary , Canada. ŌĆó Structure, Reactivity and spectra of molecules. ŌĆó Transition metal complexes and molecules with heavy atoms. 20
- 7. Computational Detail ’éĘ Structure is build from Space Group Fm-3m (no.225) ’éĘ Lattice Parameters a= 5.984Ōä½ ’éĘ Miller Indices of (001) is used to cut slab from bulk. ’éĘ Geometry Optimization ’éĘ LDA ’éĘ GGA-mPBE approximation is used. 7
Editor's Notes
- #1: DFT is a computational quantum mechanical modelling method used. DFT is used for the calculation of electric, magnetic, structural and different other properties of solids. Only up to one electron problem we can solve Schr├Čdinger's equation exactly. It is very hectic to solve the Schr├Čdinger's equation for a N- body system. In DFT instead of electronic wave function, ground state electron density Žü(r), is used to solve many body problems.
- #2: Electron density╠²is a representation of the probability of finding an╠²electron╠²in a specific location around an atom or molecule. In general, the╠²electron╠²is more likely to be found in regions with high╠²electron density. Density of electron Žü(r) only depends upon the three coordinates of position x, y and z instead of 3N-coordinates.
- #3: There are bunch of nuclei and electrons, making the equation very difficult to solve
- #4: The nuclear attraction energy part of the electronic Hamiltonian operator is called ŌĆ£external potential The part of the binding╠²energy╠²of a system of particles, such as an atomic nucleus of a solid, which is associated with electrostatic forces between the particles. Exchange correlation energy: sum of energy of interacting system. Correlation energy: interaction energy of electrons with different spin. Spin effect of electrons as well as their interaction was included.
- #5: The exchange correlation energy at any point gives the same value to that of uniform electron gas for identical density of a system. Taking the gradient of electron density improvements enhanced the accuracy of results upto large extent compared to LDA.
- #6: Amsterdam Density Functional (ADF) is particularly strong╠²in╠²understanding╠²and predicting. ADF is frequently used for studying since all elements in the periodic table can be modeled accurately
- #7: (Generalized Gradient Approximation-modified Perdew Burke Erenzerhof The lattice parameters are the quantities specifying a unit cell or the unit of the periodicity of the atomic arrangement. The lattice parameters (constants) are composed of "a, b, c," lengths of the unit cell in three dimensions, and "╬▒, ╬▓, ╬│," their mutual angles. Miller indices, group of three numbers that indicates the orientation of a plane or set of parallel planes of atoms in a crystal.