
















![Hohenberg-kohn Theorem 1
Ō¢¬ The Ground state energy E is the unique functional of
Electron Density
E = E ŃĆé [po(r)]
Ō¢¬ where p (r) represents the density function which itself is a
function of position (r).](https://image.slidesharecdn.com/densityfunctionaltheoryoverview-241215172219-46ba724c/85/Density-Functional-Theory-DFT-Overview-pptx-21-320.jpg)
![Ō¢¬ The electron density that minimizes the energy of the
overall functional is the true ground state electron density:
E[p(r)]>E.[p.(r)]
Hohenberg-kohn Theorem 2](https://image.slidesharecdn.com/densityfunctionaltheoryoverview-241215172219-46ba724c/85/Density-Functional-Theory-DFT-Overview-pptx-22-320.jpg)



































More Related Content
Similar to Density Functional Theory (DFT) Overview.pptx (20)
More from momnaqayyum01 (9)







Recently uploaded (20)




















Density Functional Theory (DFT) Overview.pptx
- 1. DENSITY FUNCTIONAL THEORY MOMNA QAYYUM Department of Chemistry University of Management and Technology, Lahore
- 2. CONTENTS Ō¢¬ DFT Ō¢¬ Why DFT Ō¢¬ H 2O Molecule Ō¢¬ Electron Density Ō¢¬ Many Particle System Ō¢¬ Born-Oppenheimer Approximations Ō¢¬ Hohenburg Kohn Theorem Ō¢¬ References
- 3. Density Functional Theory Density functional theory (DFT) is a quantum-mechanical atomistic simulation method to compute a wide variety of properties of almost any kind of atomic system: molecules, crystals, surfaces, and even electronic devices when combined with non-equilibrium Green's functions (NEGF).
- 5. Why DFT Schrodinger Equation can be solved easily for one electron system. For multiple electronic system, it is very hard to solve the Schrodinger equation. Ō¢¬ So we introduce some Approximations, one of them is DFT Ō¢¬ Reduce electron-electron interactions.
- 6. H20 Molecule
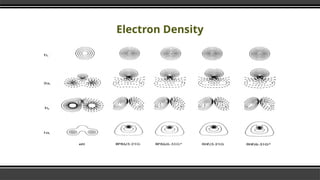
- 7. HŌééO Molecule Explained by DFT Ō¢¬ Density Functional Theory (DFT): Ō¢¬ Models the behavior of electrons in the HŌééO molecule using electron density, not wave functions. Ō¢¬ Efficiently predict molecular geometry, bond strengths, and electronic properties. Ō¢¬ Electron Density Distribution: Ō¢¬ DFT calculates the 3D electron density, showing how electrons are distributed around the oxygen and hydrogen atoms. Ō¢¬ Highlights the polar nature of the HŌééO molecule, with a higher electron density near oxygen.
- 8. HŌééO Geometry by DFT Ō¢¬ Bond Angle: ~104.5┬░ (due to lone pair repulsion on oxygen). Ō¢¬ Bond Lengths: ~0.96 ├ģ for OŌĆōH bonds (close to experimental values). Ō¢¬ Potential Energy Surface (PES): DFT maps the energy changes as the bond lengths and angles vary, providing insights into molecular stability.
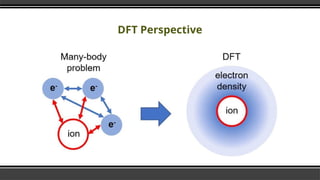
- 10. Electron Density Ō¢¬ Electron density significantly speeds up the calculation. Ō¢¬ Many body electronic wavefunction is a function of 3N variables the electron density is only a function of x, y, z only three variables. Ō¢¬ The Hohenburg-Kohn theorem asserts that the density of any system determines all ground-state properties of the system. Ō¢¬ In this case the total ground state energy of a many-electron system is a functional of the density.
- 11. Many Particle System Ō¢¬ Find the ground state for a collection of atoms by solving the Schrodinger equation Ō¢¬ ╬Ś ╬©({ri} {RI})= ╬Ģ ╬© ( {ri} {RI}) Ō¢¬ So here we have a bunch of nuclei & bunch of electron which makes a complicated equation to solve.
- 12. Born-Oppenheimer Approximations Ō¢¬ According to this approximation: Ō¢¬ Mass of nuclei is very greater than mass of electrons N>>>>e Ō¢¬ Thus nuclei are slow and electrons are fast. Ō¢¬ In other words we can write the Schrodinger wave equation by separating the electronic and nuclei terms.
- 13. Key Concepts of Born-Oppenheimer Approximations 1. Separation of Motion: Ō¢¬ Assumes that nuclei are much heavier than electrons, so their motion can be treated separately. Ō¢¬ Electrons adjust instantly to the movement of nuclei. 2. Simplifies the Schr├Čdinger Equation: Ō¢¬ Treats the electronic and nuclear wavefunctions independently, drastically reducing computational complexity. 3. Energy Surface: Ō¢¬ The nuclei move on a potential energy surface defined by the electronic energy.
- 14. Why is This Important Ō¢¬ Simplifies Molecular Calculations: Reduces a complex multi- particle system into more manageable parts. Ō¢¬ Basis for Quantum Chemistry: Enables understanding of chemical bonding and molecular dynamics. Ō¢¬ Accurate Models: Provides realistic approximations for molecular structure and spectroscopy.
- 15. Practical Applications Ō¢¬ Molecular Vibrations: Explains vibrational spectra by treating nuclei separately. Ō¢¬ Chemical Reactions: Helps map potential energy surfaces to understand reaction pathways. Ō¢¬ Spectroscopy: Provides a framework for interpreting electronic, vibrational, and rotational spectra. Ō¢¬ Molecular Dynamics Simulations: Simplifies calculations for simulating large systems. Ō¢¬ Quantum Chemistry Software: Foundational principle behind tools like Gaussian and VASP.
- 16. Hohenberg-kohn Theorem Ō¢¬ The Ground state energy E is the unique functional of Electron Density This foundational concept in Density Functional Theory (DFT) has two key parts: 1. The Ground State Density Uniquely Defines All Properties Ō¢¬ The total energy and all properties of a quantum system can be determined entirely from the electron density, not the many-electron wave function. Ō¢¬ This simplifies complex quantum calculations by reducing dimensions. 2. Existence of a Universal Energy Functional Ō¢¬ There exists a mathematical function that relates the energy of the system to the electron density. Ō¢¬ The ground-state energy corresponds to the minimum of this energy functional.
- 17. Breaking Down the Hohenberg-kohn Theorem 1. Why Focus on Electron Density? Ō¢¬ Electron density tells us where electrons are most likely to be in a molecule or material. Ō¢¬ ItŌĆÖs much simpler than the many-body wave function but still contains all the essential information. 2. Implications of the Theorem Ō¢¬ The energy and behavior of a system can be calculated without solving the complex Schr├Čdinger equation directly. Ō¢¬ This reduces the problem's complexity from many-electron interactions to just density distribution.
- 18. Key Concepts of Hohenberg-kohn Theorem Ō¢¬ The ground-state electron density uniquely determines all properties of a quantum system (e.g., energy, reactivity). Ō¢¬ Reduces complexity: Depends on 3 spatial variables, not the 3N variables of the wavefunction (N = number of electrons). Ō¢¬ A mathematical function relates the energy of the system to the electron density. Ō¢¬ The ground-state energy corresponds to the minimum of this functional. Ō¢¬ The system is in its ground state (lowest energy). Ō¢¬ The relationship between energy and density is universal for all systems.
- 19. Why is This Important Ō¢¬ Simplifies Quantum Calculations: Reduces complexity from multi-electron wavefunctions to a 3-variable electron density. Ō¢¬ Foundation of DFT: Forms the basis for Density Functional Theory, a key tool in computational chemistry and physics. Ō¢¬ Reduces Computational Cost: Enables accurate predictions for large systems with minimal computational effort. Ō¢¬ Broad Applicability: Widely used in material science, catalysis, energy storage, and drug discovery. Ō¢¬ Revolutionized Modern Chemistry: Transformed the study and design of molecules and materials with efficient modeling techniques.
- 20. Practical Applications Ō¢¬ Material design: Predicts stability, conductivity, and magnetic properties of materials. Ō¢¬ Drug Discovery: Models molecular interactions with biological targets. Ō¢¬ Catalysis: Optimizes catalysts for faster and greener reactions. Ō¢¬ Battery Development: Designs more efficient energy storage materials. Ō¢¬ Environmental Science: Models pollutant behavior and chemical reactions in the atmosphere.
- 21. Hohenberg-kohn Theorem 1 Ō¢¬ The Ground state energy E is the unique functional of Electron Density E = E ŃĆé [po(r)] Ō¢¬ where p (r) represents the density function which itself is a function of position (r).
- 22. Ō¢¬ The electron density that minimizes the energy of the overall functional is the true ground state electron density: E[p(r)]>E.[p.(r)] Hohenberg-kohn Theorem 2
- 23. From Wave function to Electron Density Ō¢¬ So, we can separate the wave function for electron and nuclei ╬©({ri} {RI})=╬©N {RI} ╬©e {ri} Ō¢¬ DFT explains the electronic density. Ō¢¬ That reduce to N dimensional to 3 spatial dimension Ō¢¬ So electron density is only 3 dimensional
- 25. References Ō¢¬ Roienko, O., Lukin, V., Oliinyk, V., Djurovi─ć, I., & Simeunovi─ć, M. (2022). An Overview of the Adaptive Robust DFT and its Applications. Technological Innovation in Engineering Research, 4, 68-89. Ō¢¬ Chakraborty, D., & Chattaraj, P. K. (2021). Conceptual density functional theory based electronic structure principles. Chemical Science, 12(18), 6264-6279. Ō¢¬ Fiechter, M. R., & Richardson, J. O. (2024). Understanding the cavity BornŌĆōOppenheimer approximation. The Journal of Chemical Physics, 160(18). Ō¢¬ Liebert, J., & Schilling, C. (2023). An exact one-particle theory of bosonic excitations: from a generalized HohenbergŌĆōKohn theorem to convexified N-representability. New Journal of Physics, 25(1), 013009.
- 26. THANK YOU