












![Kohn- Sham Approach (1965):
’āś KS replace the interacting n-electron system with a system of one-electron (non-
interacting) system in effective potential having the same ground state.
since the kinetic energy; E= Ekin+ Eext+EH +Ex+ Ec
int
non
non int
Ekin = Ekin + Ekin
where
E = Ekin + Ekin + Eext + EH +Ex + Ec
int
non int
E = Ekin + Eext + EH +Exc = F [n(r)] + Eext
non
16](https://image.slidesharecdn.com/marcomprehensiveexamondft-240520064314-2e74ea77/85/MAR_Comprehensive-exam-on-density-functional-theorypptx-16-320.jpg)



![Structural entity & stability
Table1: lattice parameter
Compound Lattice
constant
Cs2AgIrCl6 10.19
Rb2AgIrCl6 10.09
K2AgIrBr6 10.03
ŌĆó Tolerance factor
ŌĆó Octahedral factor
ŌĆó New Tolerance factor
ŌĆó Formation energy
ŌĆó Binding energy
ŌĆó Decomposition energy
ØæĪØÉ║ =
ØæģØÉ┤+ØæģØæŗ
ŌłÜ2(
ØæģØÉĄŌĆ▓+ØæģØÉĄŌĆ▓ŌĆ▓
2
+ØæģØæŗ)
[1] أŠ=
ØæģØæŗ
ØæģØÉĄ
ŌłÆ ØæøØÉ┤ ØæøØÉ┤ ŌłÆ
ØæģØÉ┤ ØæģØÉĄ
ln ØæģØÉ┤ ØæģØÉĄ
[3]
┬Ą =
ØæģØÉĄŌĆ▓+ØæģØÉĄŌĆ▓ŌĆ▓
2ØæģØæŗ
[1]
ØÉĖØæō =
ØÉĖØÉ┤2ØÉ┤ØæöØÉ╝Øæ¤ØÉČØæÖ6ŌłÆØæøØÉ┤├Ś
ØÉĖØÉ┤
Øæś
ŌłÆØæøØÉ┤Øæö├Ś
ØÉĖØÉ┤Øæö
ØæÖ
ŌłÆ ØæøØÉ╝Øæ¤├Ś
ØÉĖØÉ╝Øæ¤
ØæÜ
ŌłÆ ØæøØÉČØæÖ├Ś
ØÉĖØÉČØæÖ
ØæØ
Øæü
[2]
ØÉĖØæÅ = ØÉĖØÉ┤2ØÉ┤ØæöØÉ╝Øæ¤ØÉČØæÖ6
ŌłÆ ØæøØÉ┤ ├Ś Ø£ćØÉ┤ ŌłÆ ØæøØÉ┤Øæö ├Ś Ø£ćØÉ┤Øæö ŌłÆ ØæøØÉ╝Øæ¤ ├Ś Ø£ćØÉ╝Øæ¤ ŌłÆ ØæøØÉČØæÖ ├Ś Ø£ćØÉČØæÖ [2]
ŌłåØÉ╗ØÉĘ = 2ØÉĖ ØÉ┤ØÉČØæÖ + ØÉĖ ØÉ┤ØæöØÉČØæÖ + ØÉĖ ØÉ╝Øæ¤ØÉČØæÖ3 ŌłÆ ØÉĖ ØÉ┤2ØÉ┤ØæöØÉ╝Øæ¤ØÉČØæÖ6
1. Liu, XiangChun; Hong, Rongzi; Tian, Changsheng (24 April 2008). "Tolerance factor and the stability discussion of ABO3-type ilmenite". Journal of Materials
Science: Materials in Electronics. 20 (4): 323ŌĆō327
2. X. Du, D. He, H. Mei, Y. Zhong, and N. Cheng, ŌĆ£Insights on electronic structures, elastic features and optical properties of mixed-valence double perovskites
Cs2Au2X6 (X= F, Cl, Br, I),ŌĆØ Phys. Lett. A, vol. 384, no. 8, p. 126169, 2020.
3. C.J. Bartel, C. Sutton, B.R. Goldsmith, R.H. Ouyang, C.B. Musgrave, L. M. Ghiringhelli, M. Scheffler, New tolerance factor to predict the stability of perovskite
oxides and halides, article eaav0693, Sci. Adv. 5 (2) (2019)
20](https://image.slidesharecdn.com/marcomprehensiveexamondft-240520064314-2e74ea77/85/MAR_Comprehensive-exam-on-density-functional-theorypptx-20-320.jpg)


![Mechanical stability
C11 > 0, C11 - C12 > 0; C11 + 2C12 > 0; C44 > 0 and C11 > B > C12 [4]
Table.3: elastic parameter
Parameters Cs2AgIrCl6 Rb2AgIrCl6 K2AgIrCl6
Born
stability
127.79 90.86 78.25
35.70 19.55 17.47
16.22 18.28 12.83
92.09 71.31 60.78
199.19 129.96 113.19
19.48 1.27 4.64
Bulk modulus, B (GPa) 66.39 43.32 37.73
Shear modulus, G (GPa) 25.02 23.97 18.27
Young modulus, Y (GPa) 66.69 60.70 47.19
0.33 0.27 0.29
PughŌĆÖs ratio, B/G 2.65 1.81 2.06
0.35 0.51 0.42
4. M. Born, K. Huang, and M. Lax, ŌĆ£Dynamical theory of crystal lattices,ŌĆØ Am. J. Phys., vol. 23, no. 7, p. 474, 1955.
Y > B > G
0.26 < ductile
1.75 < ductile
23](https://image.slidesharecdn.com/marcomprehensiveexamondft-240520064314-2e74ea77/85/MAR_Comprehensive-exam-on-density-functional-theorypptx-23-320.jpg)









































More Related Content
Similar to MAR_Comprehensive exam on density functional theorypptx (20)
Recently uploaded (20)


















MAR_Comprehensive exam on density functional theorypptx
- 2. Exploration of direct band gap double perovskites A2AgIrCl6 (A= Cs, Rb, K): a DFT study Md. Abu Rayhan ID No: 20PPHY002P Session: 2020-21 Department of Physics Chittagong University of Engineering & Technology (CUET) Chittagong-4349, Bangladesh 2 Supervisor Prof. Dr. Md. Ashraf Ali
- 3. ŌĆó Introduction ŌĆó Literature review ŌĆó Motivation ŌĆó Objectives ŌĆó Methodology ŌĆó Results and discussions ŌĆó Conclusions Outlines 3
- 4. Introduction ŌĆó Perovskite materials: Crystalline compounds with unique structure (ABX3). High potential for solar cells, LEDs, due to excellent properties. Example- CaTiO3 ŌĆó Types- i) single perovskites, ii) double perovskites Single Perovskite (ABX3) ŌĆó Oxide perovskite (ABO3) ŌĆó Halide perovskite (ABF3) ŌĆó Nitrides perovskite (ABN3) Double perovskite (A2BB╩╣X6) ŌĆó Oxide ŌĆó Halide 4
- 5. Why double Perovskites Halide? ’é¦ Tunable properties ’é¦ Optoelectronic devices ’é¦ Energy conversion ’é¦ Advanced electronics ’é¦ Environmental considerations ’é¦ Magnetism and spintronics Double perovskite: A2BB╩╣X6 A= Cs+, Rb+, K+ B= Ag+ B╩╣= Ir3+ X= F-/Cl-/Br- 5
- 6. Literature review Author Compounds Property Study Journal E. Greul et al. Cs2AgBiBr6 Structural and optoelectronic J. Mater. Chem. A, vol. 5, p. 19972, 2017. N. Guechi et al. Cs2AgBiX6 (X= Cl, Br) Elastic, optoelectronic and thermoelectric J. Electron. Mater., vol. 47, pp. 1533ŌĆō1545, 2018. W. Shi et al. Cs2MBiCl6 (M= Ag, Cu, Na, K, Rb, and Cs) Structural and opto-electronic J. Chem. Phys., vol. 153, p. 141101, 2020. M. Nabi et al. Cs2CuMCl6 (M= Sb, Bi) Structural stability, electronic, elastic, thermoelectric and optical Sci. Rep., vol. 11, p. 12945, 2021. Shruthi Nair et al. Cs2TlBiI6 Structural, electronic and optical J. Phys.: Condensed Matter, vol. 31, p. 445902, 2019 T. Saha et al Cs2AgAsCl6 structural, mechanical, electronic, thermodynamic, phonon and optical Phys. Chem. Chem. Phys., vol. 24, p. 26609, 2022. T. Y. Tang et al. A2CuSbX6 (A= Cs, Rb, K; X= Cl, Br, I) Physical and optoelectronic Chemical Physics, vol. 570, p. 111897, 2023. M. Caid et al. Cs2CuIrF6 Structural stability and optoelectronic J. Molecular Modeling, vol. 29, p. 178, 2023. 6
- 7. Motivation ’ü▒ These Double perovskites compounds are (A2AgIrCl6 (A= Cs, Rb, K) composed of abundant and non-toxic elements, making them environmentally friendly along with the cost effective and time efficient. ’ü▒ No theoretical or practical work has been done on these substances in order to determine physical properties qualities via DFT computation. ’ü▒ The comprehensive study of these double perovskites also contributes to fundamental scientific understanding. 7
- 8. Objectives ’é¦ To study the structural stability such as tolerance factor, octahedral factor, new tolerance factor. ’é¦ To check the thermo-dynamical stability such as formation energy, binding energy and decomposition energy and dynamical stability of the compounds. ’é¦ To study the mechanical stability and elastic behavior. ’é¦ To find out the energy gap of band structure and electron density of states. ’é¦ To find the optical properties of the compounds for using in solar cells. ’é¦ To find the suitability for use in solar cells and/or thermoelectric devices 8
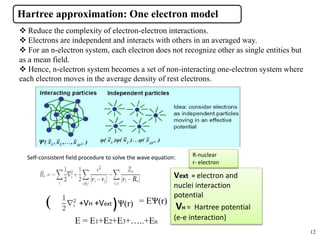
- 9. Why Density Functional Theory? 9 Computational Methodology DFT (Density Functional Theory) ’ā╝The calculation of physical and chemical properties of multi- particle systems (atoms, molecules or solids) require the exact determination of electronic structure and total energy of these systems. ’ā╝ Schr├Čdinger equation successfully explains the electronic structure of simple systems and numerically exact solutions are found for small no. of atoms and molecules. ’ā╝ This n-electron problem was solved when Kohn and Sham in 1965 formulated a theory concerning 3-dimensional electron density and energy functionals. ’ā╝Electron density n(r) plays central role instead of wave function Žł(r). The problem of many-interacting particles system in static potential is reduced to non-interacting single particle system in an effective potential.
- 10. Many body problem: ’üČ For large interacting system, we first need to consider a many particle wave function. ’üČ Many body Hamiltonian for electron and nucleus is of the form given below Hč▒ (r,R,t) =E č▒ (r,R,t) Innocent look of wave equation Hč▒ = M m e Hč▒ = = č░ = č▒ = č▒ 10
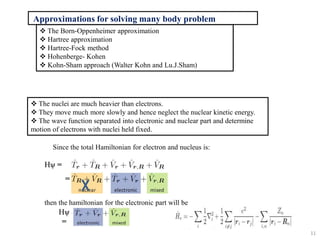
- 11. Since the total Hamiltonian for electron and nucleus is: then the hamiltonian for the electronic part will be Approximations for solving many body problem ’üČ The Born-Oppenheimer approximation ’üČ Hartree approximation ’üČ Hartree-Fock method ’üČ Hohenberge- Kohen ’üČ Kohn-Sham approach (Walter Kohn and Lu.J.Sham) ’üČ The nuclei are much heavier than electrons. ’üČ They move much more slowly and hence neglect the nuclear kinetic energy. ’üČ The wave function separated into electronic and nuclear part and determine motion of electrons with nuclei held fixed. Hč▒ = = Hč▒ = 11
- 12. Hartree approximation: One electron model ’üČ Reduce the complexity of electron-electron interactions. ’üČ Electrons are independent and interacts with others in an averaged way. ’üČ For an n-electron system, each electron does not recognize other as single entities but as a mean field. ’üČ Hence, n-electron system becomes a set of non-interacting one-electron system where each electron moves in the average density of rest electrons. Self-consistent field procedure to solve the wave equation: Vext = electron and nuclei interaction potential VH = Hartree potential (e-e interaction) ( ) +VH +Vext č░(r) = Eč░(r) E = E1+E2+E3+ŌĆ”..+En R-nuclear r- electron 12
- 13. Hartree method produced crude estimation of energy due to two oversimplifications: ’ā╝ Hartree method does not follow two basic principles of quantum mechanics: the antisymmetry principle and PauliŌĆÖs exclusion principle. ’ā╝ Does not count the exchange and correlation energies coming from n- electron nature. The Hartree method, therefore, was soon refined into the Hartree-Fock method. Hartree-Fock method Based on the one-electron and mean-field approach by Hartree, V.A. Fock enhanced the methods to higher perfection. Fock and J.C. Slater in 1930 generalized the Hartree's theory to take into account the antisymmetry requirement. ’üČ In HF method, the n-electron wave function approximated as a linear combination of non-interacting one-electron wave function in the form of Slater determinant. ’üČSlater determinant 13
- 14. VH = Vij Hartree or Coulomb interaction energy of two electrons Ex = Exchange energy comes from the antisymmetric nature of wave function in the Slater determinant. Difficulties with Hartree-Fock Theory: A new approach has been developed known as Density Functional Theory (DFT). ’āś In 1964 Hohenberg and Kohn showed that schrodinger equation (3N dimensional e.g. 10 electrons require 30 dimensions) could be reformulated in terms of electron density n(r) with non-interacting n separate 3-dimensional ones. ’āś The main objective of DFT is to replace the many-particle electronic wavefunction with the electron density as the basic quantity. ’āś The electron density n(r), the central player in DFT decides everything in an n-electron quantum state where there is no individual electron density but a 3-dimensional density of electrons. ’āś The addition of all the electron densities over the whole space naturally return to the total number of electrons in the system. ’āś The knowledge of overlapping of atomic electron density, roughly generate the electron density of the solids. ’āś This theory gives approximate solutions to both Exchange and Correlation Energies. ’āś Correlation energy and ’āś Problem of dealing 3N dimensional . )č▒(r) = E č▒(r) E = Ekin+ EH +Eext + Ex 14
- 15. The Fundamental Pillars of DFT First Hohenberg Kohn (HK) theorem: The ground-state energy is a unique functional of the electron density n(r). ’āś This theorem provides one to one mapping between ground state wave function and ground state charge density. ’āś The ground state charge density can uniquely describe all the ground state properties of system. ’āś The fundamental concept behind density functional theory is that charge density (3- Dimensional) can correctly describe the ground state of N-particle instead of using a wave function (3N-Dimensional). Second Hohenberg Kohn (HK) theorem: The electron density that minimizes the energy of the overall functional is the true electron density. ’āś If the true functional form of energy in terms of density gets known, then one could vary the electron density until the energy from the functional is minimized, giving us required ground state density. ’āś This is essentially a variational principle and is used in practice with approximate forms of the functional. ’āś The simplest possible choice of a functional can be a constant electron density all over the space. 15
- 16. Kohn- Sham Approach (1965): ’āś KS replace the interacting n-electron system with a system of one-electron (non- interacting) system in effective potential having the same ground state. since the kinetic energy; E= Ekin+ Eext+EH +Ex+ Ec int non non int Ekin = Ekin + Ekin where E = Ekin + Ekin + Eext + EH +Ex + Ec int non int E = Ekin + Eext + EH +Exc = F [n(r)] + Eext non 16
- 17. Hence final KH equation has the form: DFT in Practice: Kohn-Sham Self Consistency loop 17
- 18. 1. Local density approximation (LDA) Exchange-correlation approximation ’āś Approximation used to find out exchange- correlation function. ’āś Exchange-correlation energy functional is purely local. ’āś Ignores corrections to the exchange- correlation energy at a point r due to nearby inhomogeneities in the electron density. 2. Generalized Gradient Approximation (GGA) ’āś Depends on local density and its gradient. ’āś GGA uses information about the local electron density and also the local gradient in the electron density. Though GGA includes more physical information than LDA. It is not necessary that it must be more accurate. There are large number of distinct GGA functionals depending on the ways in which information from the gradient of the electron density can be included in a GGA functional. 18
- 19. Results & Discussion Structural parameter ’é¦ Cubic structure ’é¦ Space group (Fm-3m, 225) ’é¦ Lattice parameter (a = b = c; ╬▒ = ╬▓ = ╬│ = 90Ōü░) ’é¦ Cs/Rb/K (0.25, 0.25, 0.25), Ag(0.5, 0.5, 0.5), Ir (0, 0, 0), and Cl (0.25, 0, 0). ’é¦ Four formula unit (2:1:1:6) Fig.2: Unit cell of Cs2AgIrCl6 19
- 20. Structural entity & stability Table1: lattice parameter Compound Lattice constant Cs2AgIrCl6 10.19 Rb2AgIrCl6 10.09 K2AgIrBr6 10.03 ŌĆó Tolerance factor ŌĆó Octahedral factor ŌĆó New Tolerance factor ŌĆó Formation energy ŌĆó Binding energy ŌĆó Decomposition energy ØæĪØÉ║ = ØæģØÉ┤+ØæģØæŗ ŌłÜ2( ØæģØÉĄŌĆ▓+ØæģØÉĄŌĆ▓ŌĆ▓ 2 +ØæģØæŗ) [1] أŠ= ØæģØæŗ ØæģØÉĄ ŌłÆ ØæøØÉ┤ ØæøØÉ┤ ŌłÆ ØæģØÉ┤ ØæģØÉĄ ln ØæģØÉ┤ ØæģØÉĄ [3] ┬Ą = ØæģØÉĄŌĆ▓+ØæģØÉĄŌĆ▓ŌĆ▓ 2ØæģØæŗ [1] ØÉĖØæō = ØÉĖØÉ┤2ØÉ┤ØæöØÉ╝Øæ¤ØÉČØæÖ6ŌłÆØæøØÉ┤├Ś ØÉĖØÉ┤ Øæś ŌłÆØæøØÉ┤Øæö├Ś ØÉĖØÉ┤Øæö ØæÖ ŌłÆ ØæøØÉ╝Øæ¤├Ś ØÉĖØÉ╝Øæ¤ ØæÜ ŌłÆ ØæøØÉČØæÖ├Ś ØÉĖØÉČØæÖ ØæØ Øæü [2] ØÉĖØæÅ = ØÉĖØÉ┤2ØÉ┤ØæöØÉ╝Øæ¤ØÉČØæÖ6 ŌłÆ ØæøØÉ┤ ├Ś Ø£ćØÉ┤ ŌłÆ ØæøØÉ┤Øæö ├Ś Ø£ćØÉ┤Øæö ŌłÆ ØæøØÉ╝Øæ¤ ├Ś Ø£ćØÉ╝Øæ¤ ŌłÆ ØæøØÉČØæÖ ├Ś Ø£ćØÉČØæÖ [2] ŌłåØÉ╗ØÉĘ = 2ØÉĖ ØÉ┤ØÉČØæÖ + ØÉĖ ØÉ┤ØæöØÉČØæÖ + ØÉĖ ØÉ╝Øæ¤ØÉČØæÖ3 ŌłÆ ØÉĖ ØÉ┤2ØÉ┤ØæöØÉ╝Øæ¤ØÉČØæÖ6 1. Liu, XiangChun; Hong, Rongzi; Tian, Changsheng (24 April 2008). "Tolerance factor and the stability discussion of ABO3-type ilmenite". Journal of Materials Science: Materials in Electronics. 20 (4): 323ŌĆō327 2. X. Du, D. He, H. Mei, Y. Zhong, and N. Cheng, ŌĆ£Insights on electronic structures, elastic features and optical properties of mixed-valence double perovskites Cs2Au2X6 (X= F, Cl, Br, I),ŌĆØ Phys. Lett. A, vol. 384, no. 8, p. 126169, 2020. 3. C.J. Bartel, C. Sutton, B.R. Goldsmith, R.H. Ouyang, C.B. Musgrave, L. M. Ghiringhelli, M. Scheffler, New tolerance factor to predict the stability of perovskite oxides and halides, article eaav0693, Sci. Adv. 5 (2) (2019) 20
- 21. Table.2: Different stability factor DP Ionic radius of cations (Ōä½) Ionic radius of anion (Ōä½) Tolerance factor (t) Octahedral factor (ØØü) New tolerance factor (ØØē) Binding energy, Øæ¼ØÆā (eV/atom) Formation enthalpy, Øæ¼ØÆć (eV/atom) Decomp osition energy, ŌłåØÉ╗ØÉĘ (meV/at om) Cs2AgIrCl6 Øæ¤ØÉČØæĀ 1.88 (Øæ¤ØÉ┤Øæö+Øæ¤ØÉ╝Øæ¤)/2 0.92 Øæ¤ØÉČØæÖ 1.81 0.96 0.51 3.83 -4.29 -2.10 73.55 Rb2AgIrCl6 Øæ¤Rb 1.72 (Øæ¤ØÉ┤Øæö+Øæ¤ØÉ╝Øæ¤)/2 0.92 Øæ¤ØÉČØæÖ 1.81 0.91 0.51 3.93 -4.28 -2.06 50.66 Cs2AgIrBr6 Øæ¤K 1.64 (Øæ¤ØÉ┤Øæö+Øæ¤ØÉ╝Øæ¤)/2 0.92 Øæ¤ØÉČØæÖ 1.81 0.89 0.51 4.04 -4.28 -2.03 0.19 0.81 Ōēż ØæĪ Ōēż 1.1; 0.41 Ōēż ┬Ą Ōēż 0.89; Žä < 4.18 21
- 22. Dynamical stability Fig.3: Phonon dispersion with total and partial DOS of A2AgIrCl6 ; A= Cs, Rb, K 22
- 23. Mechanical stability C11 > 0, C11 - C12 > 0; C11 + 2C12 > 0; C44 > 0 and C11 > B > C12 [4] Table.3: elastic parameter Parameters Cs2AgIrCl6 Rb2AgIrCl6 K2AgIrCl6 Born stability 127.79 90.86 78.25 35.70 19.55 17.47 16.22 18.28 12.83 92.09 71.31 60.78 199.19 129.96 113.19 19.48 1.27 4.64 Bulk modulus, B (GPa) 66.39 43.32 37.73 Shear modulus, G (GPa) 25.02 23.97 18.27 Young modulus, Y (GPa) 66.69 60.70 47.19 0.33 0.27 0.29 PughŌĆÖs ratio, B/G 2.65 1.81 2.06 0.35 0.51 0.42 4. M. Born, K. Huang, and M. Lax, ŌĆ£Dynamical theory of crystal lattices,ŌĆØ Am. J. Phys., vol. 23, no. 7, p. 474, 1955. Y > B > G 0.26 < ductile 1.75 < ductile 23
- 24. Electronic properties ŌĆó Band structure ŌĆó DOS (TDOS and PDOS) ŌĆó Direct band gap nature ŌĆó Effective mass of electrons and holes Table.4: Band gap values of compounds Compounds Band gap (eV) Functional Band gap nature Effective mass of electron, ØÆÄØÆå ŌłŚ Effective mass of hole, ØÆÄØÆē ŌłŚ Cs2AgIrCl6 0.34 1.43 GGA PBE Tb-mBJ Direct Direct 0.13 me 0.19 me 1.10 me 1.43 me Rb2AgIrCl6 0.36 1.50 GGA PBE Tb-mBJ Direct Direct 0.13 me 0.18 me 1.10 me 1.70 me K2AgIrCl6 0.38 1.55 GGA PBE Tb-mBJ Direct Direct 0.13 me 0.18 me 1.64 me 1.14 me 24
- 25. Fig. 4: Energy band structure of Cs2AgIrCl6, Rb2AgIrCl6, K2AgIrCl6 by GGA and TB-mBJ method. 25
- 26. Fig 5: Total and partial DOS of Cs2AgIrCl6, Rb2AgIrCl6, K2AgIrCl6 by TB-mBJ method 26
- 27. Fig. 6 Charge density of A2AgIrCl6 (A = Cs, Rb, K) compounds 27
- 28. Optical properties Fig.7: Real and imaginary part of dielectric constant, refractive index, extinction coefficient of the studied compound 28
- 29. Optical properties Fig.8: Absorption coefficient, optical conductivity, reflectivity, and loss function of the studied compound 29
- 31. Fig. 9 Calculated thermoelectric properties including a) Electrical conductivity, b) Electronic portion of thermal conductivity, c) Lattice thermal conductivity, d) Seebeck coefficient, e) Power factor (PF), and f) Figure of merit (ZT) of Cs2AgIrCl6 (red color), Rb2AgIrCl6 (blue color), and K2AgIrCl6 (green color) double perovskite. 31
- 32. Table 5: The calculated values of electrical conductivity (Žā), electronic conductivity (Ke), Seebeck coefficient (S), power factor (PF), and figure of merit (ZT) for A2AgIrCl6 (A=Cs, Rb, K) at room temperature. Material property Cs2AgIrCl6 Rb2AgIrCl6 K2AgIrCl6 Transport properties (300 K) Ø£Ä ├Ś 105 ╬® ŌłÖ m 0.66 0.67 0.68 Ke (Wm-1K-1) 0.97 0.85 0.91 S (╬╝V/K) 189.30 176.23 182.20 PF (├Ś10-3 Wm-1K-2) 2.40 2.09 2.25 ZT 0.74 0.74 0.83 32
- 33. Conclusions: ŌĆó The suggested compounds exhibit stability in dynamic, thermodynamic, and mechanical aspects. ŌĆó The compounds exhibit a characteristic of ductility. ŌĆó The A2AgIrCl6 possesses a direct band gap. ŌĆó The energy gap corresponds to the visible range of electromagnetic waves, making it applicable for utilization in solar cells, renewable energy technology, and photocatalytic applications. ŌĆó Effective mass of electrons are small compared to effective mass of holes indicating higher carrier mobility. ŌĆó The high absorption coefficients of 105 order and other optical constant, making their suitability for opto-electronic application. ŌĆó The low reflectivity values (less than 13%) also indicates their high absorption ability. ŌĆó A2AgIrCl6 (A = Cs/Rb/K) are desirable choice for potential candidates for use in thermoelectric devices. 33
- 34. THANK YOU! 34