












































More Related Content
What's hot (20)
Viewers also liked (17)















Similar to Gen pop4ld (20)




















More from Genetica, Ferrara University, Italy (15)















Gen pop4ld
- 1. Genetica di popolazioni 4: Linkage disequilibrium
- 2. Programma del corso 1. Diversità genetica 2. Equilibrio di Hardy-Weinberg 3. Inbreeding 4. Linkage disequilibrium 5. Mutazione 6. Deriva genetica 7. Flusso genico e varianze genetiche 8. Selezione 9. Mantenimento dei polimorfismi e teoria neutrale 10. Introduzione alla teoria coalescente 11. Struttura e storia della popolazione umana + Lettura critica di articoli
- 3. Linkage e linkage disequilibrium Linkage: l’associazione fisica dei loci sui cromosomi, Linkage disequilibrium: l’associazione non casuale degli alleli di diversi loci nei cromosomi/gameti, a formare aplotipi. Il linkage è una causa (non la sola) del linkage disequilibrum
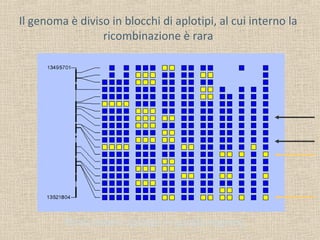
- 5. Il genoma è diviso in blocchi di aplotipi, al cui interno la ricombinazione è rara Nove diversi aplotipi in questa regione
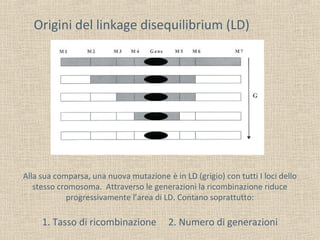
- 6. Origini del linkage disequilibrium (LD) Alla sua comparsa, una nuova mutazione è in LD (grigio) con tutti I loci dello stesso cromosoma. Attraverso le generazioni la ricombinazione riduce progressivamente l’area di LD. Contano soprattutto: 1. Tasso di ricombinazione 2. Numero di generazioni
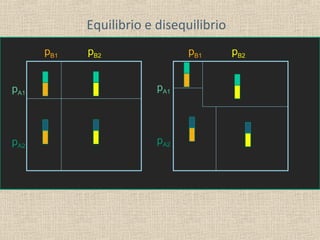
- 7. Quindi: Equilibrio di Hardy-Weinberg e linkage disequilibrium • Basta una generazione di accoppiamento casuale per raggiungere l’equilibrio di HW a un locus • Se si studiano più loci, possono essere necessarie parecchie generazioni perché si raggiunga anche un linkage equilibrium, cioé perché gli alleli siano associati casualmente nei gameti
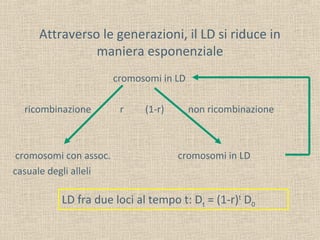
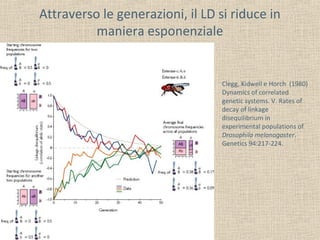
- 8. Attraverso le generazioni, il LD si riduce in maniera esponenziale cromosomi in LD ricombinazione cromosomi con assoc. casuale degli alleli r (1-r) non ricombinazione cromosomi in LD LD fra due loci al tempo t: Dt = (1-r)t D0
- 9. Attraverso le generazioni, il LD si riduce in maniera esponenziale Clegg, Kidwell e Horch (1980) Dynamics of correlated genetic systems. V. Rates of decay of linkage disequilibrium in experimental populations of Drosophila melanogaster. Genetics 94:217-224.
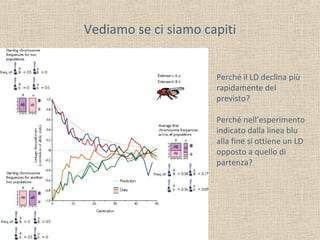
- 10. Vediamo se ci siamo capiti Perché il LD declina più rapidamente del previsto? Perché nell’esperimento indicato dalla linea blu alla fine si ottiene un LD opposto a quello di partenza?
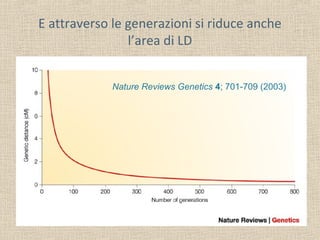
- 11. E attraverso le generazioni si riduce anche l’area di LD Nature Reviews Genetics 4; 701-709 (2003)
- 12. Se gli alleli ai due loci non sono associati in maniera casuale, ci sarà una deviazione delle frequenze degli aplotipi (D) rispetto alle frequenze attese: p11 = p1q1 + D p12 = p1q2 – D p21 = p2q1 – D p22 = p2q2 + D Il parametro D è il coefficiente di linkage disequilibrium, proposto per primi da Lewontin e Kojima (1960). Dmax è uguale a min (p1q2, p2q1) per D>0, o a max (-p1q1, -p2q2) per D < 0.
- 14. Il valore assoluto di D dipende dalle frequenze alleliche, il suo segno dalle etichette arbitrarie attribuite agli alleli (1 o 2) D = p11 – pA1pB1 = p11p22 – p12p21 Se pA1= pB1 = 0.50 Dmax = 0.50 - 0.25 = 0.25 (p11=0.5, p22=0.5) Dmin = 0.00 - 0.25 = -0.25 (p12=0.5, p21=0.5) Se pA1≠ 0.50 o pB1 ≠ 0.50, p. es. 0.20 e 0.80 Dmax = 0.20 - 0.16 = 0.04 (p11=0.2, p22=0.2, p12=0.6) Dmin = 0.00 - 0.16 = -0.16 (p12=0.8, p21=0.2)
- 19. Un sito web che calcola LD http://www.evotutor.org/EvoGen/EG4A.html
- 20. Non tutti i geni sono trasmessi indipendentemente, perché ci sono più loci che cromosomi da Kidd et al. 1999
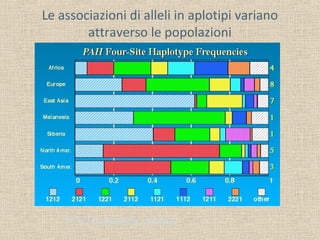
- 21. Le associazioni di alleli in aplotipi variano attraverso le popolazioni 24 = 16 possibili aplotipi
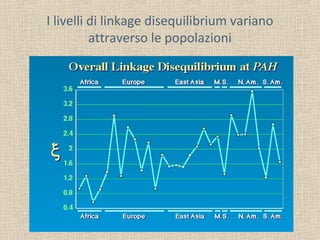
- 22. I livelli di linkage disequilibrium variano attraverso le popolazioni
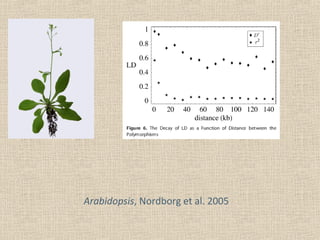
- 23. Arabidopsis, Nordborg et al. 2005
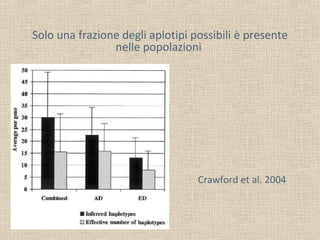
- 24. Solo una frazione degli aplotipi possibili è presente nelle popolazioni Crawford et al. 2004
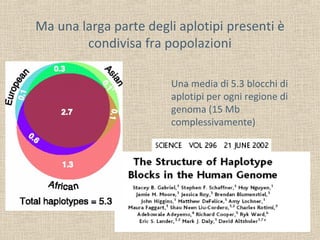
- 25. Ma una larga parte degli aplotipi presenti è condivisa fra popolazioni Una media di 5.3 blocchi di aplotipi per ogni regione di genoma (15 Mb complessivamente)
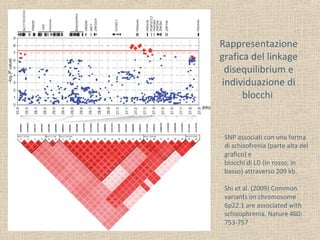
- 26. Rappresentazione grafica del linkage disequilibrium e individuazione di blocchi SNP associati con una forma di schizofrenia (parte alta del grafico) e blocchi di LD (in rosso, in basso) attraverso 209 kb. Shi et al. (2009) Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature 460: 753-757
- 27. Una review non recentissima, ma ancora buona
- 28. Sintesi • Due loci sono in linkage equilibrium se le frequenze genotipiche a un locus sono indipendenti da quelle all’altro locus • Il linkage disequilibrium è causato dalla mutazione e ridotto dalla ricombinazione • Basta una generazione di accoppiamento casuale per raggiungere l’equilibrio di Hardy-Weinberg, ma non il LD • Si misura il LD confrontando frequenze genotipiche osservate e attese a due loci, tramite le statistiche r, D e D’